Department of Genetics and Genomic Science, Icahn School of Medicine at Mount Sinai, New York, NY, 10029, USA.
Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY, 10029, USA.
Genome Biol. 2020 Dec 8;21(1):298. doi: 10.1186/s13059-020-02194-x.
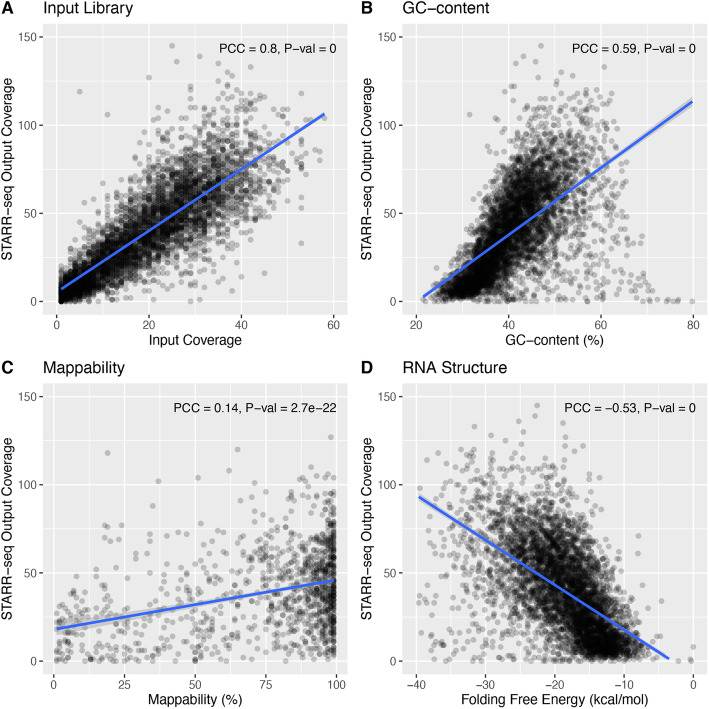
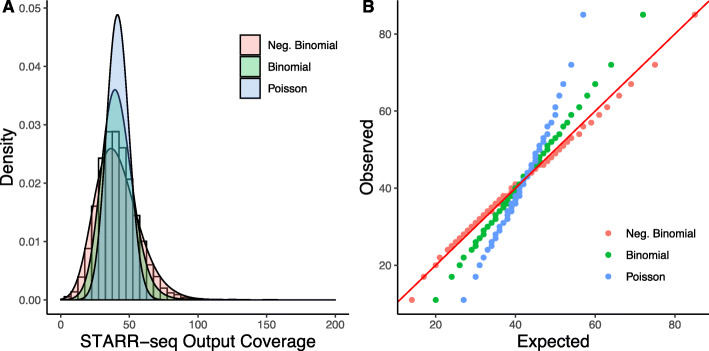
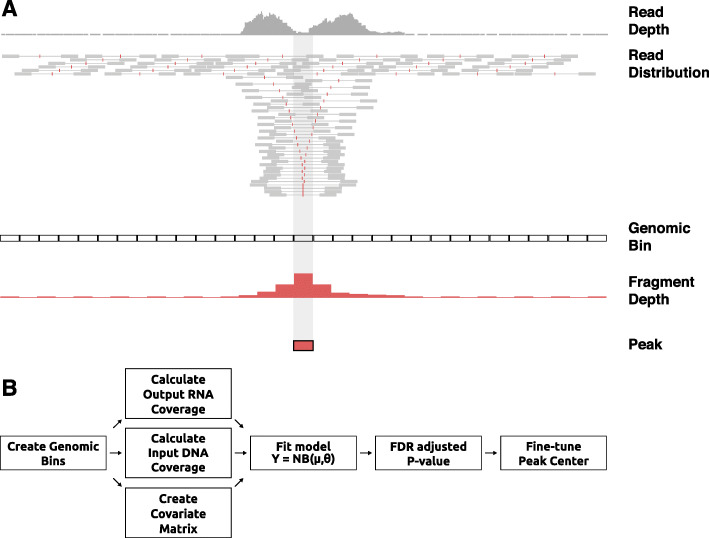
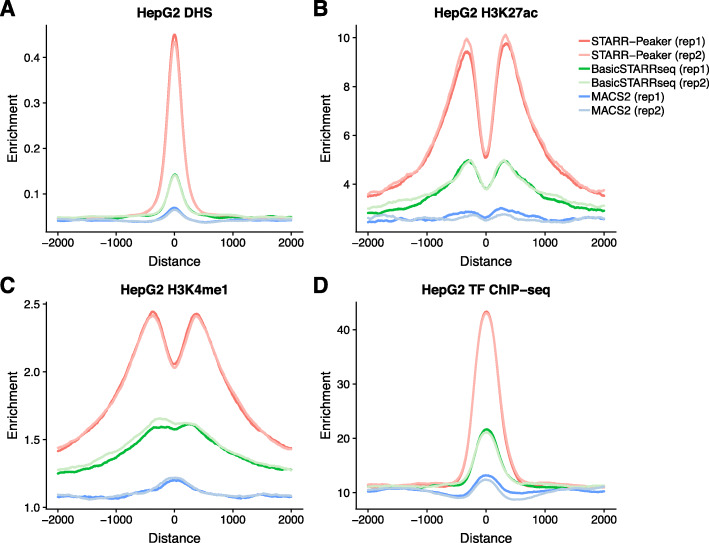
STARR-seq technology has employed progressively more complex genomic libraries and increased sequencing depths. An issue with the increased complexity and depth is that the coverage in STARR-seq experiments is non-uniform, overdispersed, and often confounded by sequencing biases, such as GC content. Furthermore, STARR-seq readout is confounded by RNA secondary structure and thermodynamic stability. To address these potential confounders, we developed a negative binomial regression framework for uniformly processing STARR-seq data, called STARRPeaker. Moreover, to aid our effort, we generated whole-genome STARR-seq data from the HepG2 and K562 human cell lines and applied STARRPeaker to comprehensively and unbiasedly call enhancers in them.
STARR-seq 技术采用了越来越复杂的基因组文库和增加的测序深度。增加的复杂性和深度的一个问题是,STARR-seq 实验中的覆盖度是非均匀的、过度分散的,并且经常受到测序偏差(如 GC 含量)的影响。此外,STARR-seq 的读取受到 RNA 二级结构和热力学稳定性的影响。为了解决这些潜在的混杂因素,我们开发了一种负二项式回归框架,用于统一处理 STARR-seq 数据,称为 STARRPeaker。此外,为了帮助我们的研究,我们从 HepG2 和 K562 人类细胞系中生成了全基因组 STARR-seq 数据,并应用 STARRPeaker 全面且无偏地对它们进行增强子的调用。