School of Mathematics, Renmin University of China, Beijing, 100872, People's Republic of China.
Department of Cryptography and Technology, Beijing Electronic Science and Technology Institute, Beijing, 100070, People's Republic of China.
Sci Rep. 2020 Dec 10;10(1):21773. doi: 10.1038/s41598-020-78465-1.
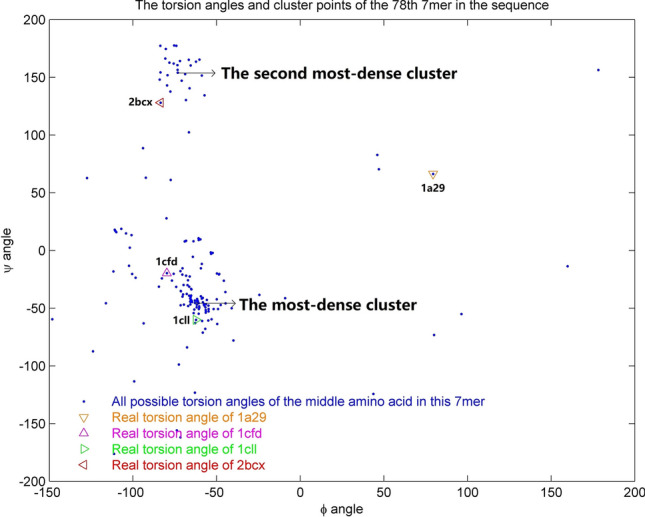
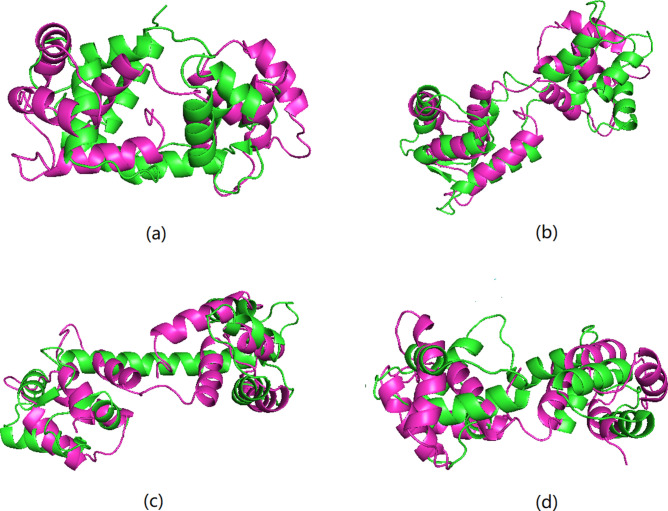
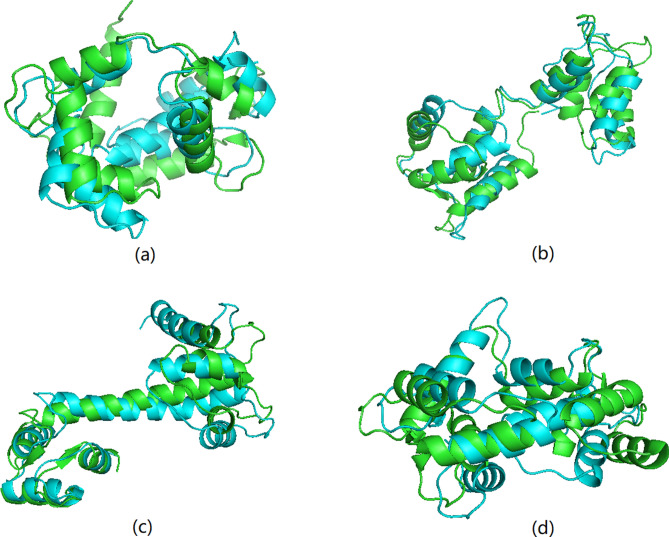
Protein structure can provide insights that help biologists to predict and understand protein functions and interactions. However, the number of known protein structures has not kept pace with the number of protein sequences determined by high-throughput sequencing. Current techniques used to determine the structure of proteins are complex and require a lot of time to analyze the experimental results, especially for large protein molecules. The limitations of these methods have motivated us to create a new approach for protein structure prediction. Here we describe a new approach to predict of protein structures and structure classes from amino acid sequences. Our prediction model performs well in comparison with previous methods when applied to the structural classification of two CATH datasets with more than 5000 protein domains. The average accuracy is 92.5% for structure classification, which is higher than that of previous research. We also used our model to predict four known protein structures with a single amino acid sequence, while many other existing methods could only obtain one possible structure for a given sequence. The results show that our method provides a new effective and reliable tool for protein structure prediction research.
蛋白质结构可以提供帮助生物学家预测和理解蛋白质功能和相互作用的见解。然而,已知的蛋白质结构数量并没有跟上高通量测序所确定的蛋白质序列数量的步伐。目前用于确定蛋白质结构的技术很复杂,需要大量时间来分析实验结果,特别是对于大型蛋白质分子。这些方法的局限性促使我们开发了一种新的蛋白质结构预测方法。在这里,我们描述了一种从氨基酸序列预测蛋白质结构和结构类别的新方法。与以前的方法相比,我们的预测模型在应用于两个包含 5000 多个蛋白质结构域的 CATH 数据集的结构分类时表现良好。结构分类的平均准确率为 92.5%,高于以往的研究。我们还使用我们的模型预测了四个具有单个氨基酸序列的已知蛋白质结构,而许多其他现有方法只能为给定序列获得一个可能的结构。结果表明,我们的方法为蛋白质结构预测研究提供了一种新的有效可靠的工具。