Appel Institute for Alzheimer's Disease Research, Brain and Mind Research Institute, Weill Cornell Medicine, New York, NY, USA.
EMBO Mol Med. 2021 Jan 11;13(1):e12354. doi: 10.15252/emmm.202012354. Epub 2020 Dec 17.
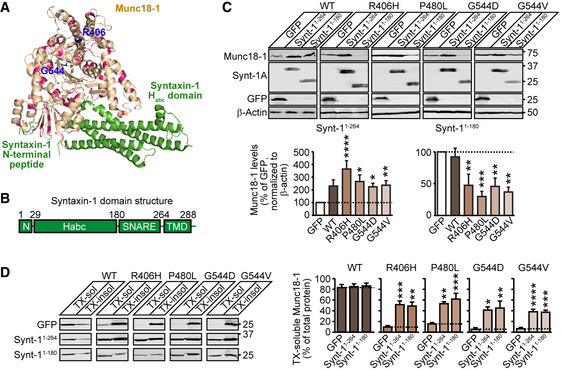
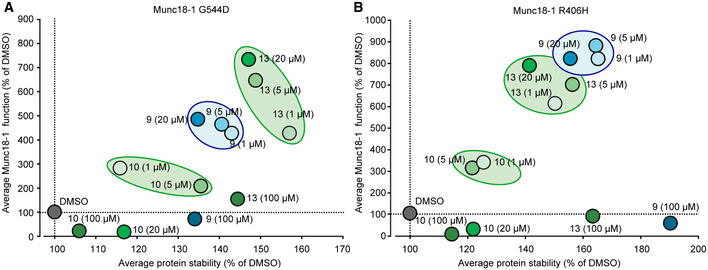
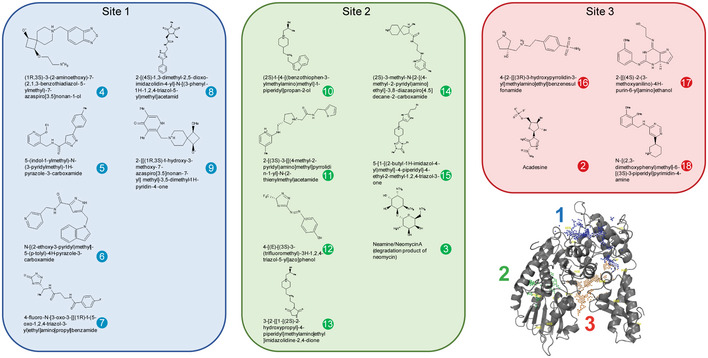
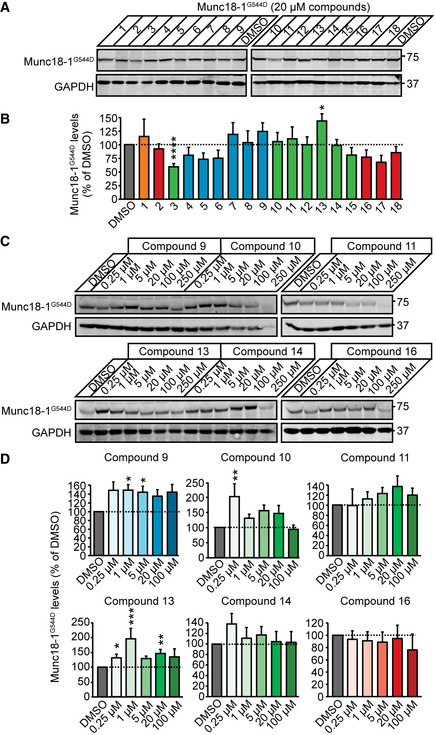
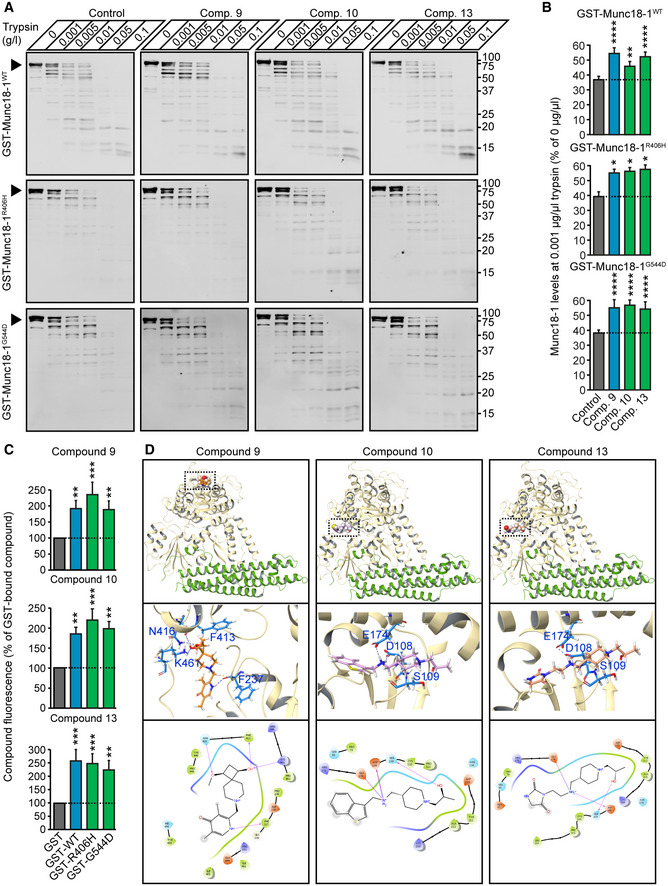
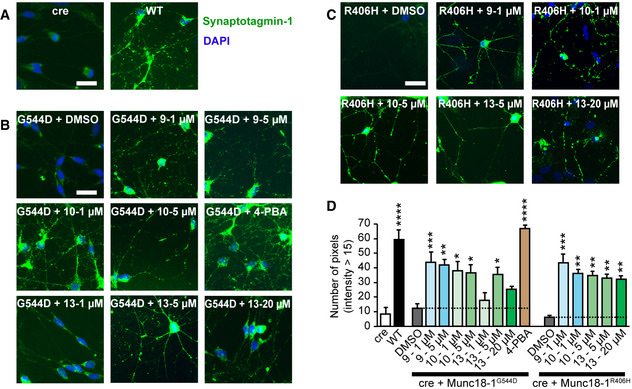
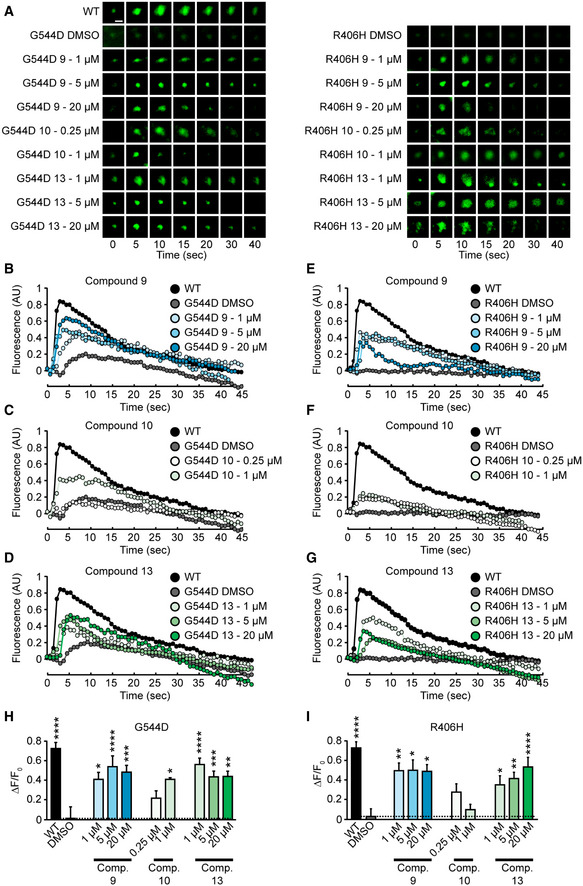
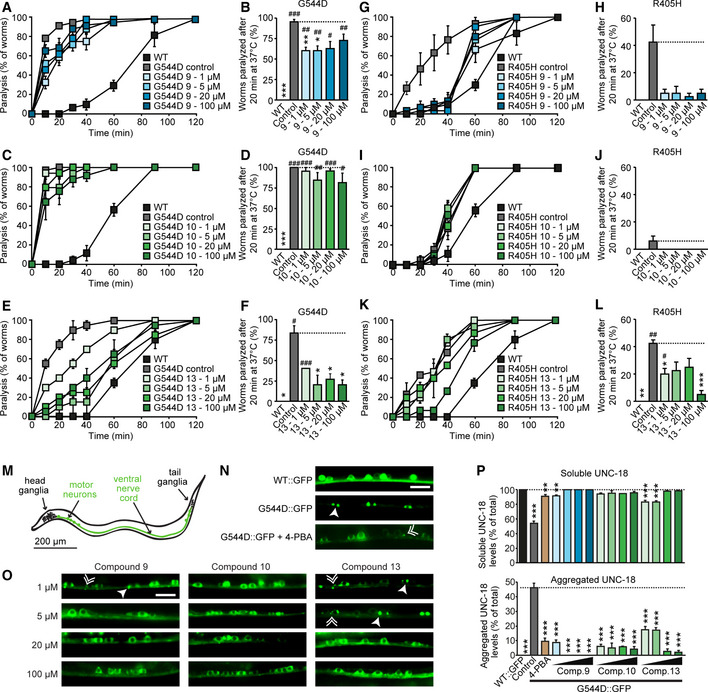
Heterozygous de novo mutations in the neuronal protein Munc18-1 cause syndromic neurological symptoms, including severe epilepsy, intellectual disability, developmental delay, ataxia, and tremor. No disease-modifying therapy exists to treat these disorders, and while chemical chaperones have been shown to alleviate neuronal dysfunction caused by missense mutations in Munc18-1, their required high concentrations and potential toxicity necessitate a Munc18-1-targeted therapy. Munc18-1 is essential for neurotransmitter release, and mutations in Munc18-1 have been shown to cause neuronal dysfunction via aggregation and co-aggregation of the wild-type protein, reducing functional Munc18-1 levels well below hemizygous levels. Here, we identify two pharmacological chaperones via structure-based drug design, that bind to wild-type and mutant Munc18-1, and revert Munc18-1 aggregation and neuronal dysfunction in vitro and in vivo, providing the first targeted treatment strategy for these severe pediatric encephalopathies.
神经元蛋白 Munc18-1 的杂合性新生突变会导致综合征性神经症状,包括严重的癫痫、智力残疾、发育迟缓、共济失调和震颤。目前尚无治疗这些疾病的方法,虽然化学伴侣已被证明可以减轻 Munc18-1 错义突变引起的神经元功能障碍,但它们所需的高浓度和潜在毒性需要一种针对 Munc18-1 的治疗方法。Munc18-1 对神经递质释放至关重要,并且 Munc18-1 的突变已被证明通过野生型蛋白的聚集和共聚集导致神经元功能障碍,使功能性 Munc18-1 水平降低到低于半合子水平。在这里,我们通过基于结构的药物设计确定了两种药理学伴侣,它们与野生型和突变型 Munc18-1 结合,并在体外和体内逆转 Munc18-1 聚集和神经元功能障碍,为这些严重的小儿脑病提供了首个靶向治疗策略。