DIFFER-Dutch Institute for Fundamental Energy Research, De Zaale 20, 5612 AJ, Eindhoven, The Netherlands.
CCER-Center for Computational Energy Research, De Zaale 20, 5612 AJ, Eindhoven, The Netherlands.
Sci Rep. 2020 Dec 17;10(1):22149. doi: 10.1038/s41598-020-79153-w.
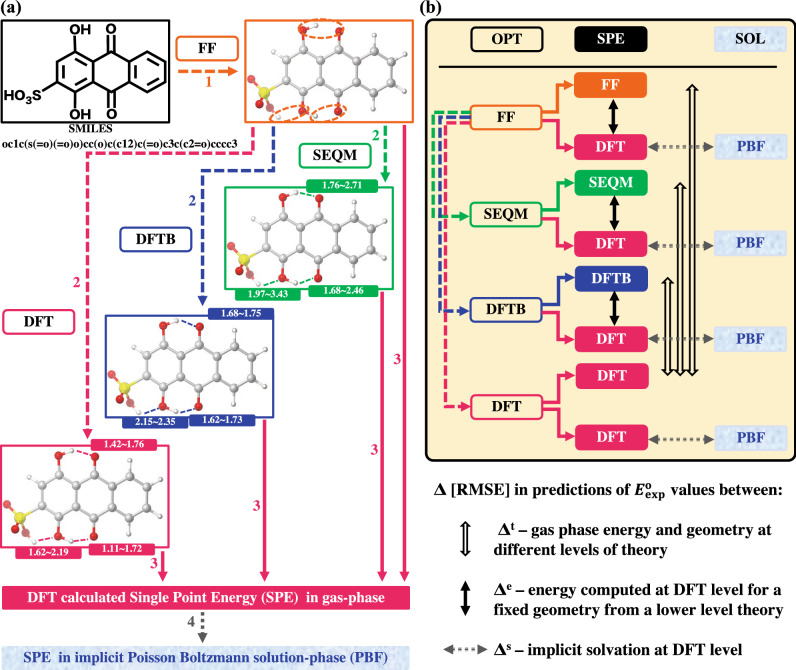
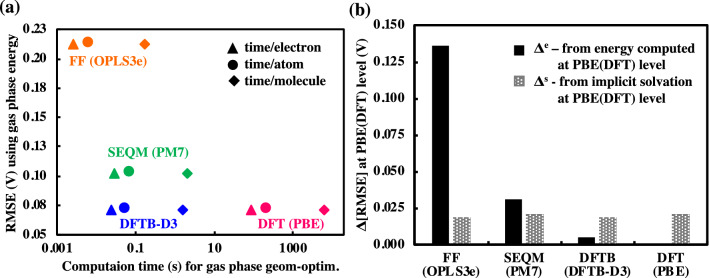
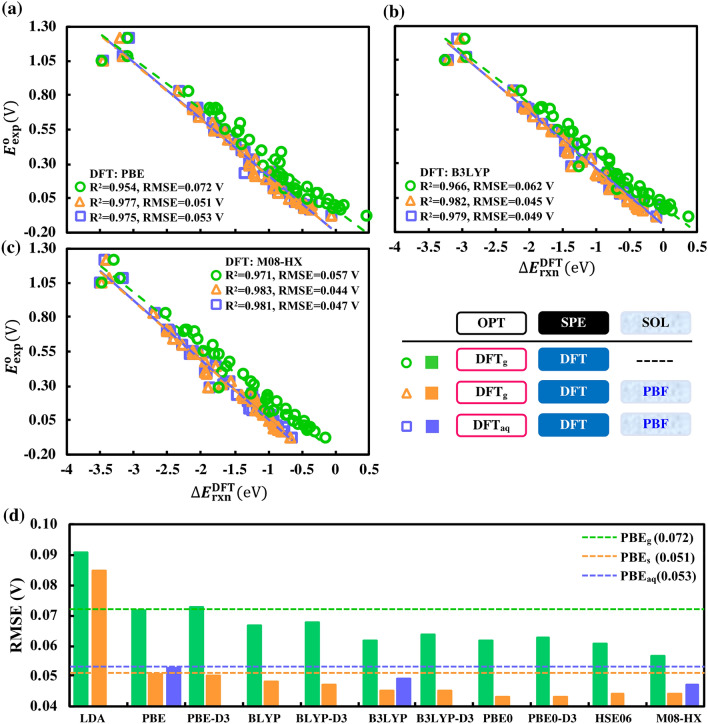
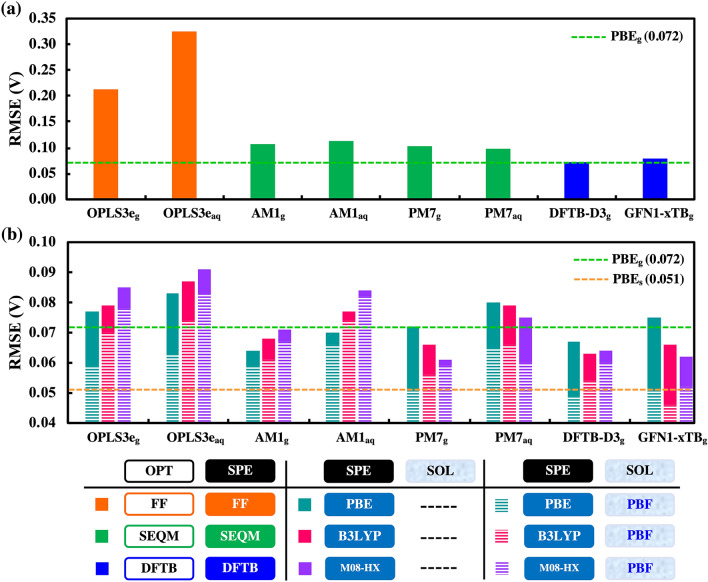
High-throughput computational screening (HTCS) is a powerful approach for the rational and time-efficient design of electroactive compounds. The effectiveness of HTCS is dependent on accuracy and speed at which the performance descriptors can be estimated for possibly millions of candidate compounds. Here, a systematic evaluation of computational methods, including force field (FF), semi-empirical quantum mechanics (SEQM), density functional based tight binding (DFTB), and density functional theory (DFT), is performed on the basis of their accuracy in predicting the redox potentials of redox-active organic compounds. Geometry optimizations at low-level theories followed by single point energy (SPE) DFT calculations that include an implicit solvation model are found to offer equipollent accuracy as the high-level DFT methods, albeit at significantly lower computational costs. Effects of implicit solvation on molecular geometries and SPEs, and their overall effects on the prediction accuracy of redox potentials are analyzed in view of computational cost versus prediction accuracy, which outlines the best choice of methods corresponding to a desired level of accuracy. The modular computational approach is applicable for accelerating the virtual studies on functional quinones and the respective discovery of candidate compounds for energy storage.
高通量计算筛选 (HTCS) 是一种用于合理且高效地设计电活性化合物的强大方法。HTCS 的有效性取决于可以为可能数百万种候选化合物估算性能描述符的准确性和速度。在这里,根据它们预测氧化还原活性有机化合物氧化还原电位的准确性,对包括力场 (FF)、半经验量子力学 (SEQM)、基于密度泛函的紧束缚 (DFTB) 和密度泛函理论 (DFT) 在内的计算方法进行了系统评估。在低水平理论上进行几何优化,然后进行单点能量 (SPE) DFT 计算,其中包括隐式溶剂化模型,被发现与高水平 DFT 方法具有等效的准确性,尽管计算成本要低得多。从计算成本与预测准确性的角度分析了隐式溶剂化对分子几何形状和 SPE 的影响,以及它们对氧化还原电位预测准确性的总体影响,从而概述了根据所需精度选择方法的最佳选择。模块化计算方法适用于加速功能醌类的虚拟研究以及储能候选化合物的发现。