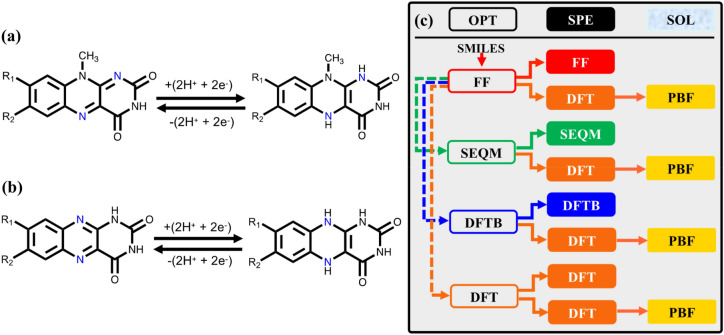
Zhang Qi, Khetan Abhishek, Er Süleyman
DIFFER-Dutch Institute for Fundamental Energy Research, De Zaale 20, 5612 AJ, Eindhoven, The Netherlands.
CCER-Center for Computational Energy Research, De Zaale 20, 5612 AJ, Eindhoven, The Netherlands.
Sci Rep. 2021 Feb 18;11(1):4089. doi: 10.1038/s41598-021-83605-2.
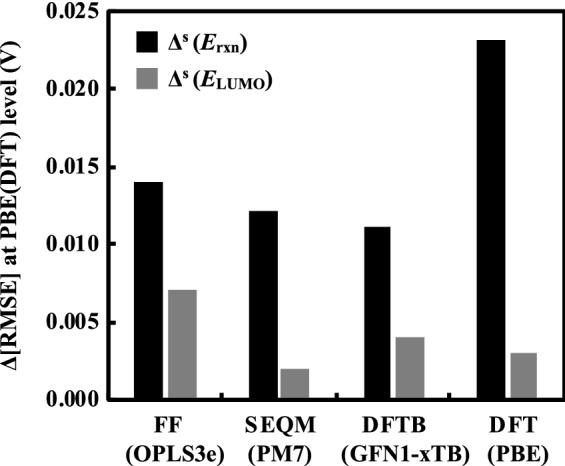
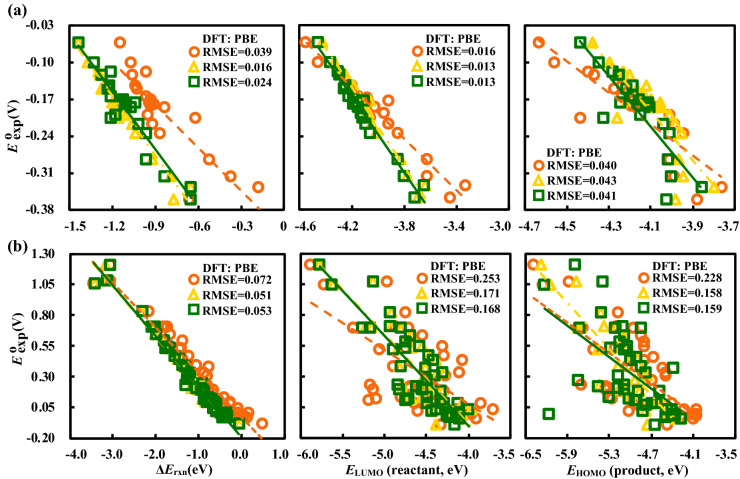
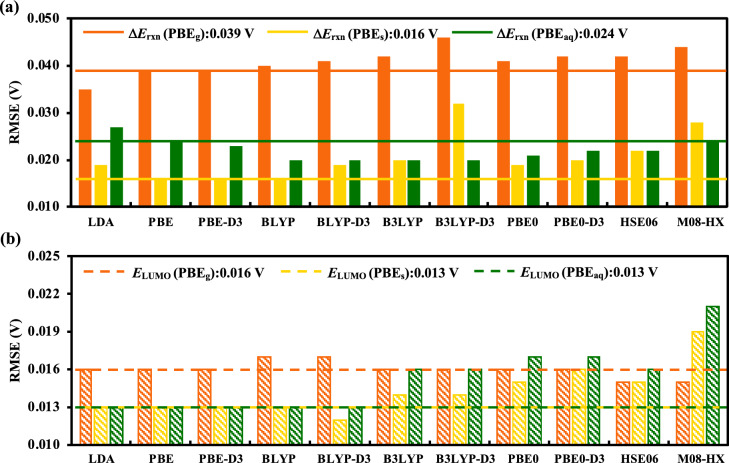
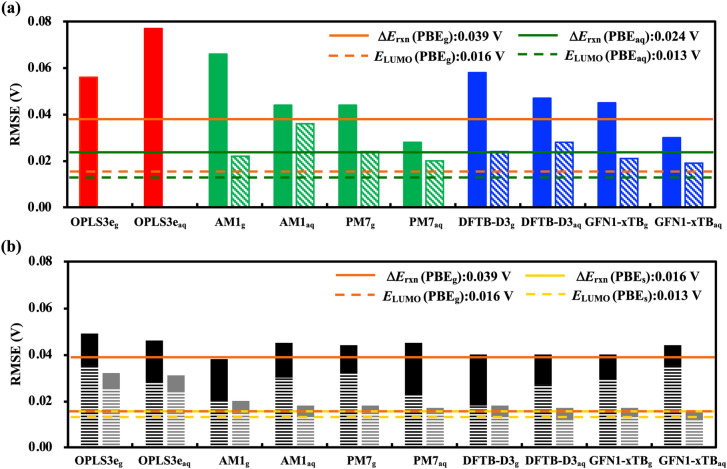
Alloxazines are a promising class of organic electroactive compounds for application in aqueous redox flow batteries (ARFBs), whose redox properties need to be tuned further for higher performance. High-throughput computational screening (HTCS) enables rational and time-efficient study of energy storage compounds. We compared the performance of computational chemistry methods, including the force field based molecular mechanics, semi-empirical quantum mechanics, density functional tight binding, and density functional theory, on the basis of their accuracy and computational cost in predicting the redox potentials of alloxazines. Various energy-based descriptors, including the redox reaction energies and the frontier orbital energies of the reactant and product molecules, were considered. We found that the lowest unoccupied molecular orbital (LUMO) energy of the reactant molecules is the best performing chemical descriptor for alloxazines, which is in contrast to other classes of energy storage compounds, such as quinones that we reported earlier. Notably, we present a flexible in silico approach to accelerate both the singly and the HTCS studies, therewithal considering the level of accuracy versus measured electrochemical data, which is readily applicable for the discovery of alloxazine-derived organic compounds for energy storage in ARFBs.
咯嗪是一类很有前景的有机电活性化合物,可应用于水系氧化还原液流电池(ARFBs),但其氧化还原性质还需要进一步调整以实现更高的性能。高通量计算筛选(HTCS)能够对储能化合物进行合理且高效的研究。我们基于计算化学方法在预测咯嗪氧化还原电位时的准确性和计算成本,比较了包括基于力场的分子力学、半经验量子力学、密度泛函紧束缚和密度泛函理论在内的多种方法。考虑了各种基于能量的描述符,包括氧化还原反应能量以及反应物和产物分子的前沿轨道能量。我们发现反应物分子的最低未占据分子轨道(LUMO)能量是咯嗪表现最佳的化学描述符,这与我们之前报道的其他类储能化合物(如醌类)不同。值得注意的是,我们提出了一种灵活的计算机模拟方法来加速单体系和高通量计算筛选研究,同时考虑与实测电化学数据相比的准确性水平,该方法很容易应用于发现用于ARFBs储能的咯嗪衍生有机化合物。