Wanchai Visanu, Nookaew Intawat, Ussery David W
Arkansas Center for Genomic Epidemiology & Medicine and The Department of Biomedical Informatics, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA.
Comput Struct Biotechnol J. 2020 Nov 24;18:3890-3896. doi: 10.1016/j.csbj.2020.10.023. eCollection 2020.
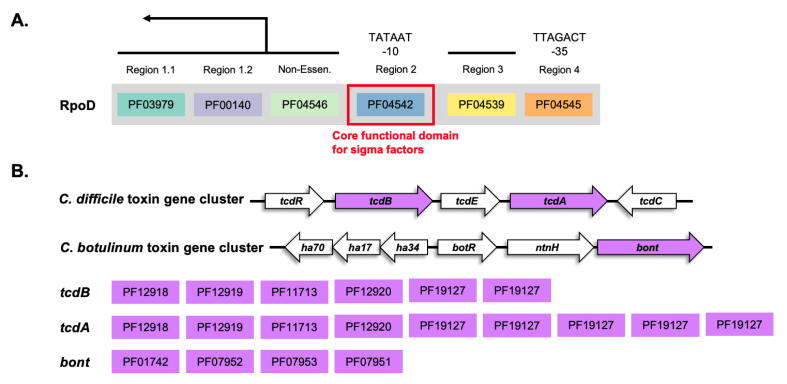
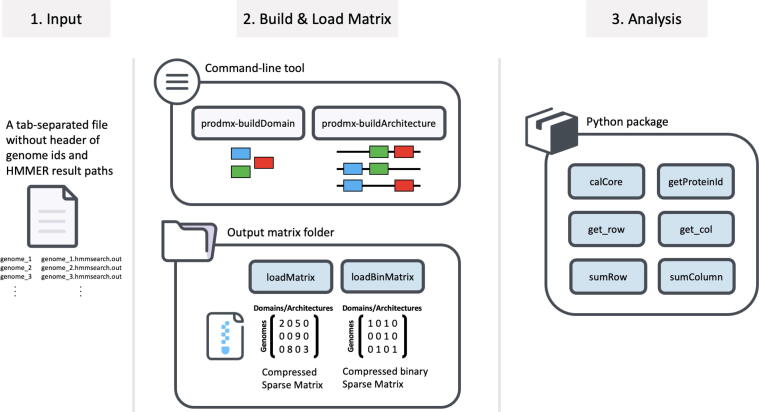
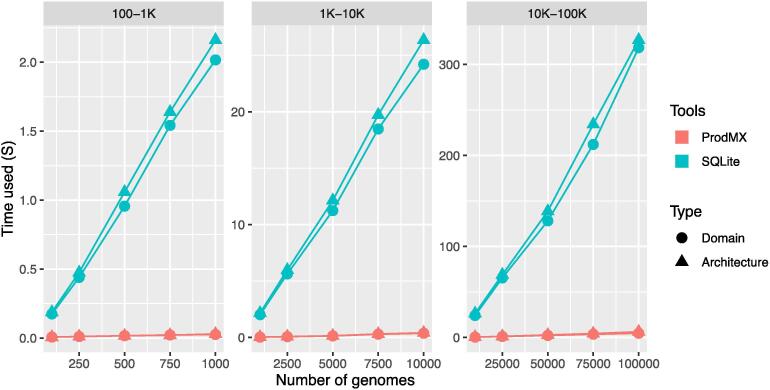
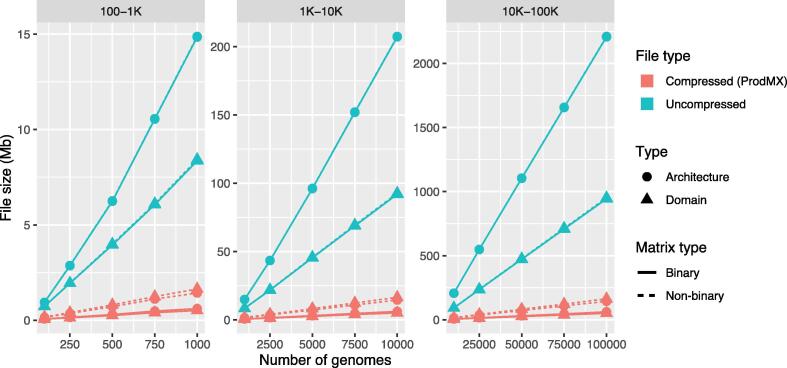
Large-scale protein analysis has been used to characterize large numbers of proteins across numerous species. One of the applications is to use as a high-throughput screening method for pathogenicity of genomes. Unlike sequence homology methods, protein comparison at a functional level provides us with a unique opportunity to classify proteins, based on their functional structures without dealing with sequence complexity of distantly related species. Protein functions can be abstractly described by a set of protein functional domains, such as PfamA domains; a set of genomes can then be mapped to a matrix, with each row representing a genome, and the columns representing the presence or absence of a given functional domain. However, a powerful tool is needed to analyze the large sparse matrices generated by millions of genomes that will become available in the near future. The ProdMX is a tool with user-friendly utilities developed to facilitate high-throughput analysis of proteins with an ability to be included as an effective module in the high-throughput pipeline. The ProdMX employs a compressed sparse matrix algorithm to reduce computational resources and time used to perform the matrix manipulation during functional domain analysis. The ProdMX is a free and publicly available Python package which can be installed with popular package mangers such as PyPI and Conda, or with a standard installer from source code available on the ProdMX GitHub repository at https://github.com/visanuwan/prodmx.
大规模蛋白质分析已被用于表征众多物种中的大量蛋白质。其中一个应用是用作基因组致病性的高通量筛选方法。与序列同源性方法不同,在功能水平上进行蛋白质比较为我们提供了一个独特的机会,可根据蛋白质的功能结构对其进行分类,而无需处理远缘物种的序列复杂性。蛋白质功能可以通过一组蛋白质功能域(如PfamA结构域)进行抽象描述;然后可以将一组基因组映射到一个矩阵,其中每行代表一个基因组,列代表给定功能域的存在或不存在。然而,需要一个强大的工具来分析由数百万个基因组生成的大型稀疏矩阵,这些矩阵在不久的将来将会出现。ProdMX是一个具有用户友好实用程序的工具,开发用于促进蛋白质的高通量分析,并能够作为高通量流程中的一个有效模块。ProdMX采用压缩稀疏矩阵算法来减少在功能域分析期间执行矩阵操作所使用的计算资源和时间。ProdMX是一个免费的、公开可用的Python包,可以使用流行的包管理器(如PyPI和Conda)进行安装,也可以使用来自https://github.com/visanuwan/prodmx上ProdMX GitHub存储库的源代码通过标准安装程序进行安装。