Bioinformatics Laboratory, Department of Statistics, University of Rajshahi, Rajshahi, Bangladesh.
Department of Microbiology, Rajshahi Institute of Biosciences, University of Rajshahi, Rajshahi, Bangladesh.
PLoS One. 2020 Dec 21;15(12):e0228233. doi: 10.1371/journal.pone.0228233. eCollection 2020.
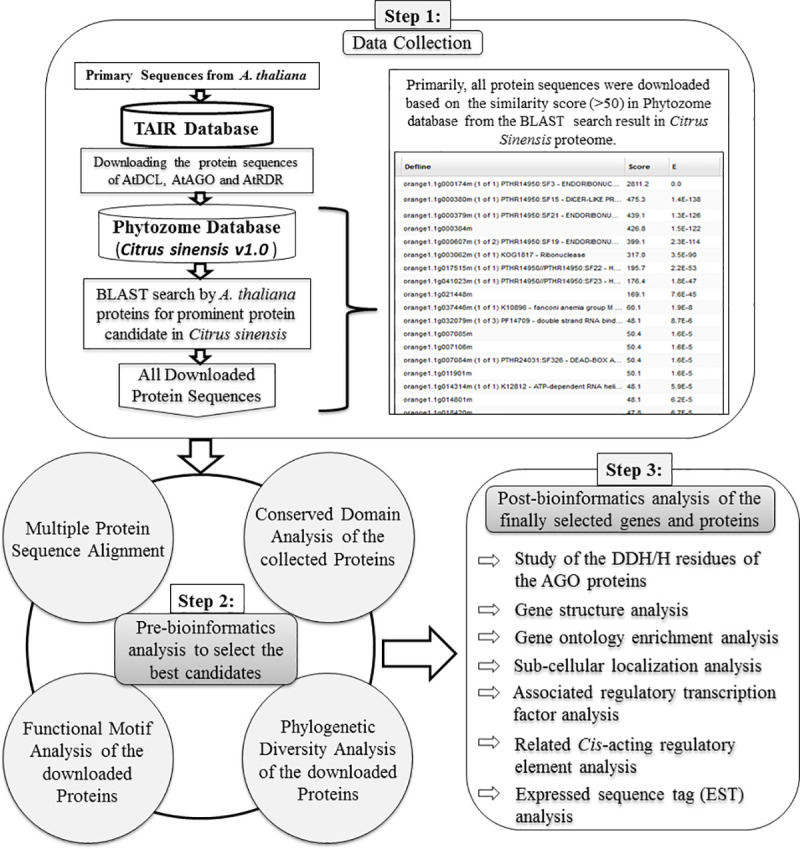
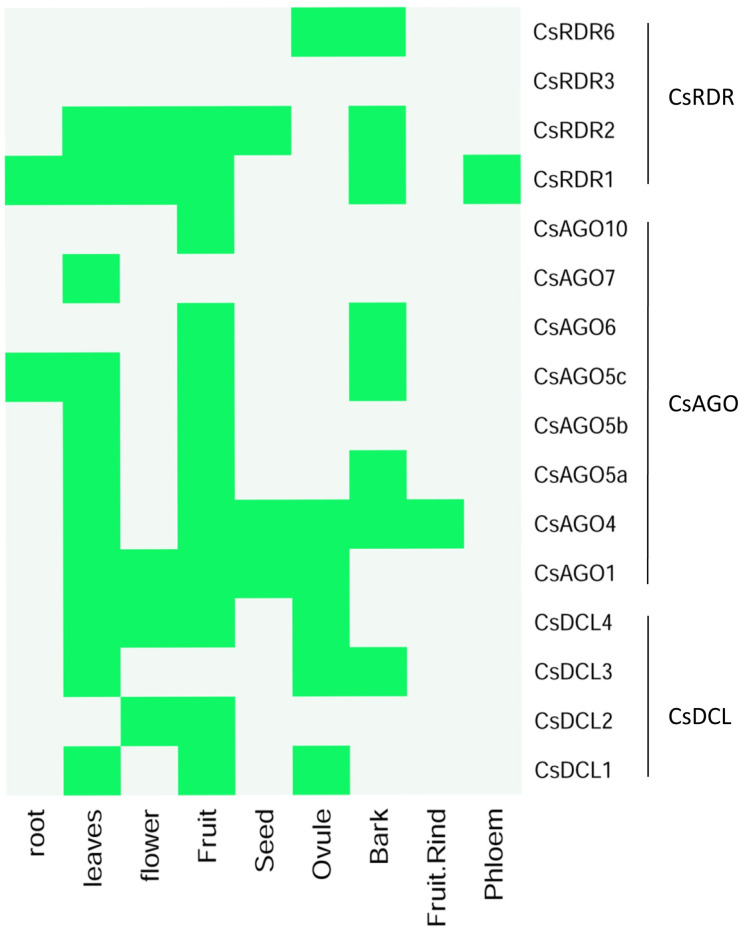
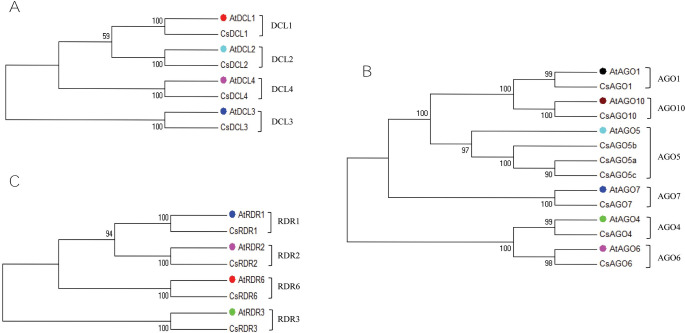
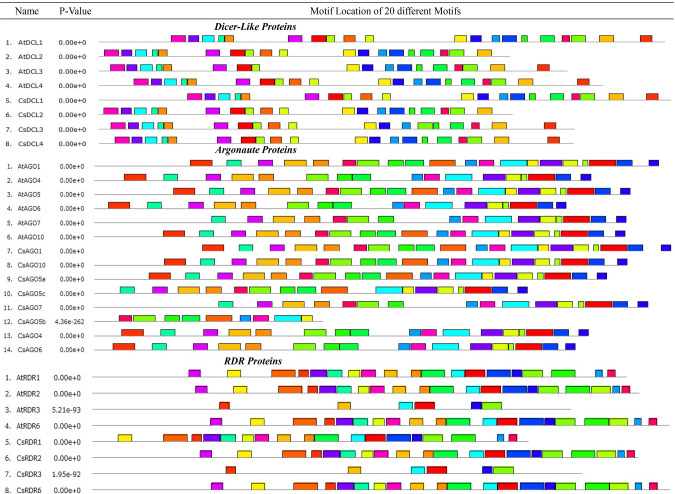
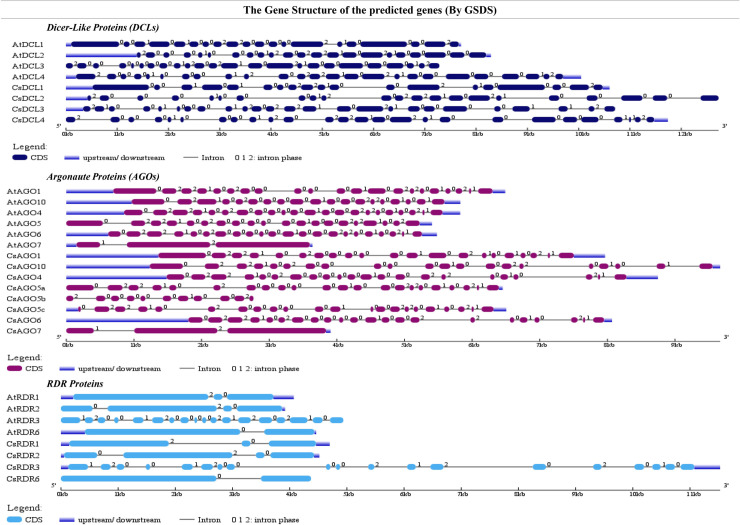
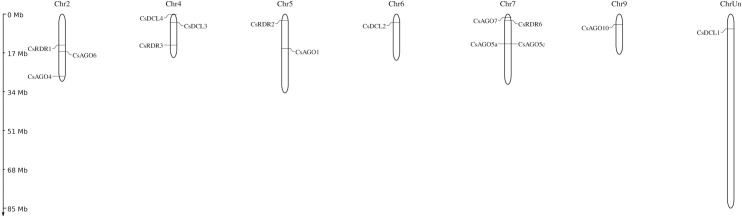
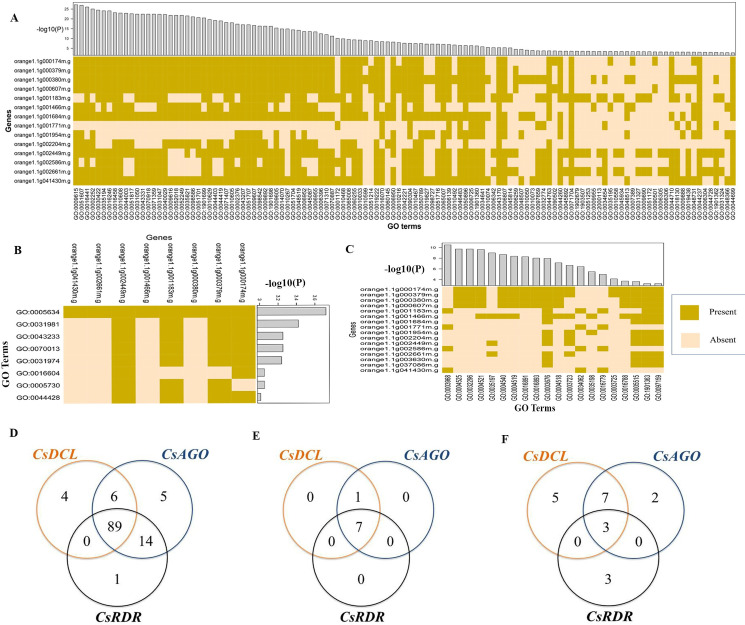
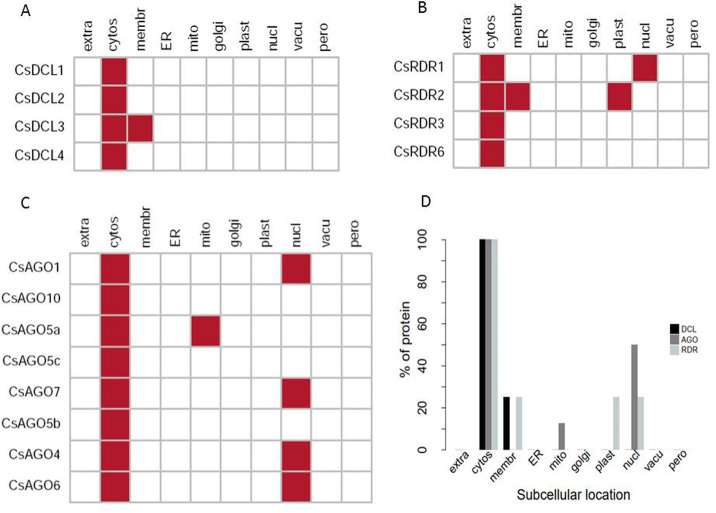
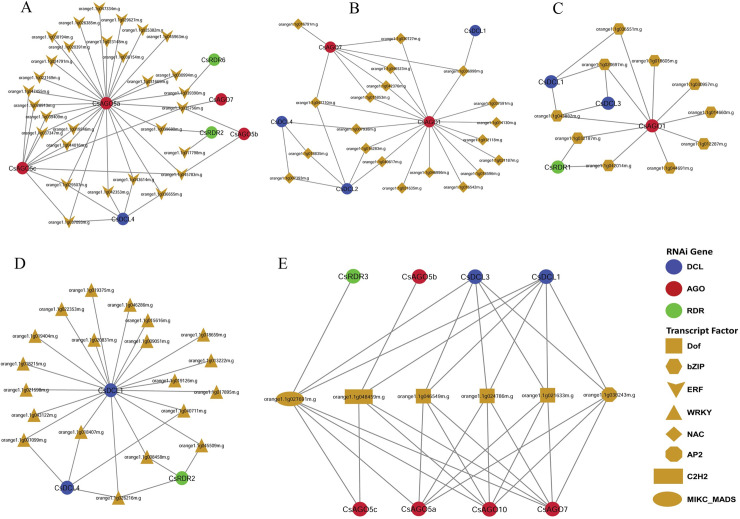
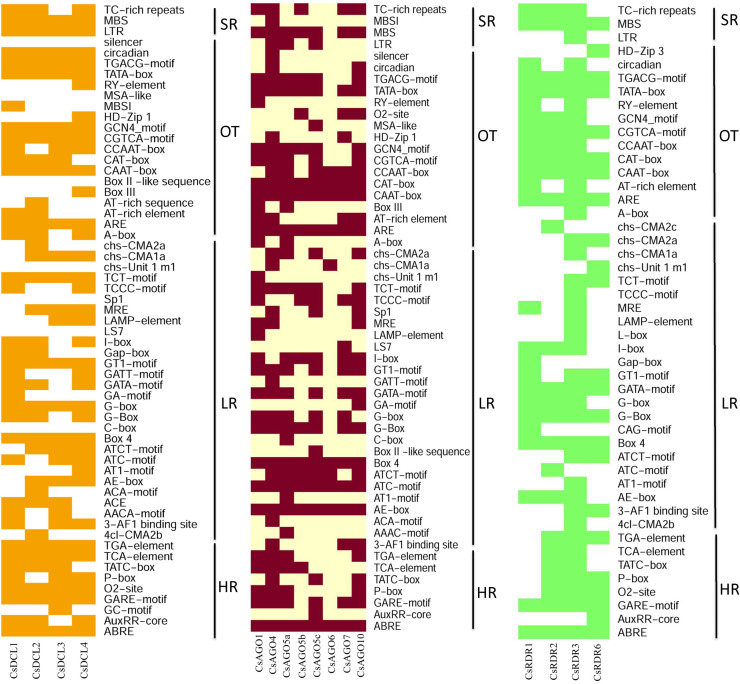
RNA interference (RNAi) plays key roles in post-transcriptional and chromatin modification levels as well as regulates various eukaryotic gene expressions which are involved in stress responses, development and maintenance of genome integrity during developmental stages. The whole mechanism of RNAi pathway is directly involved with the gene-silencing process by the interaction of Dicer-Like (DCL), Argonaute (AGO) and RNA-dependent RNA polymerase (RDR) gene families and their regulatory elements. However, these RNAi gene families and their sub-cellular locations, functional pathways and regulatory components were not extensively investigated in the case of economically and nutritionally important fruit plant sweet orange (Citrus sinensis L.). Therefore, in silico characterization, gene diversity and regulatory factor analysis of RNA silencing genes in C. sinensis were conducted by using the integrated bioinformatics approaches. Genome-wide comparison analysis based on phylogenetic tree approach detected 4 CsDCL, 8 CsAGO and 4 CsRDR as RNAi candidate genes in C. sinensis corresponding to the RNAi genes of model plant Arabidopsis thaliana. The domain and motif composition and gene structure analyses for all three gene families exhibited almost homogeneity within the same group members. The Gene Ontology enrichment analysis clearly indicated that the predicted genes have direct involvement into the gene-silencing and other important pathways. The key regulatory transcription factors (TFs) MYB, Dof, ERF, NAC, MIKC_MADS, WRKY and bZIP were identified by their interaction network analysis with the predicted genes. The cis-acting regulatory elements associated with the predicted genes were detected as responsive to light, stress and hormone functions. Furthermore, the expressed sequence tag (EST) analysis showed that these RNAi candidate genes were highly expressed in fruit and leaves indicating their organ specific functions. Our genome-wide comparison and integrated bioinformatics analyses provided some necessary information about sweet orange RNA silencing components that would pave a ground for further investigation of functional mechanism of the predicted genes and their regulatory factors.
RNA 干扰 (RNAi) 在转录后和染色质修饰水平上发挥着关键作用,同时调节各种真核基因的表达,这些基因参与应激反应、发育以及基因组完整性的维持。RNAi 途径的整个机制直接涉及基因沉默过程,通过 Dicer-Like (DCL)、Argonaute (AGO) 和 RNA 依赖性 RNA 聚合酶 (RDR) 基因家族及其调控元件的相互作用。然而,在经济和营养上重要的水果植物甜橙 (Citrus sinensis L.) 中,这些 RNAi 基因家族及其亚细胞位置、功能途径和调控成分并没有得到广泛研究。因此,本研究通过整合生物信息学方法,对甜橙中的 RNA 沉默基因进行了基因多样性和调控因子分析。基于系统发育树方法的全基因组比较分析,在甜橙中检测到 4 个 CsDCL、8 个 CsAGO 和 4 个 CsRDR,作为与拟南芥 RNAi 基因相对应的 RNAi 候选基因。所有三个基因家族的结构域和基序组成以及基因结构分析表明,同一组内的成员几乎具有同质性。GO 富集分析清楚地表明,预测基因直接参与基因沉默和其他重要途径。通过与预测基因的互作网络分析,鉴定出关键的调控转录因子 (TF) MYB、Dof、ERF、NAC、MIKC_MADS、WRKY 和 bZIP。与预测基因相关的顺式作用调控元件被检测到对光、胁迫和激素功能有反应。此外,表达序列标签 (EST) 分析表明,这些 RNAi 候选基因在果实和叶片中高度表达,表明它们具有器官特异性功能。本研究的全基因组比较和整合生物信息学分析为甜橙 RNA 沉默成分提供了一些必要的信息,为进一步研究预测基因及其调控因子的功能机制奠定了基础。