Di Meo Ivano, Cavestro Chiara, Pedretti Silvia, Fu Tingting, Ligorio Simona, Manocchio Antonello, Lavermicocca Lucrezia, Santambrogio Paolo, Ripamonti Maddalena, Levi Sonia, Ayciriex Sophie, Mitro Nico, Tiranti Valeria
Unit of Medical Genetics and Neurogenetics, Fondazione IRCCS Istituto Neurologico Carlo Besta, 20126 Milan, Italy.
DiSFeB, Dipartimento di Scienze Farmacologiche e Biomolecolari, Università degli Studi di Milano, 20133 Milan, Italy.
Int J Mol Sci. 2020 Dec 19;21(24):9707. doi: 10.3390/ijms21249707.
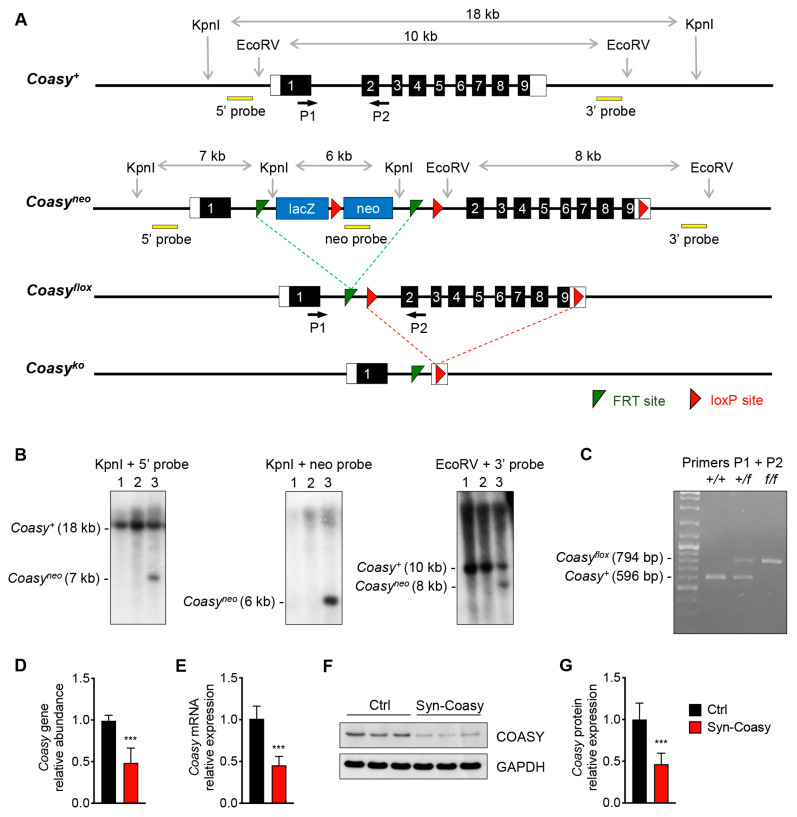
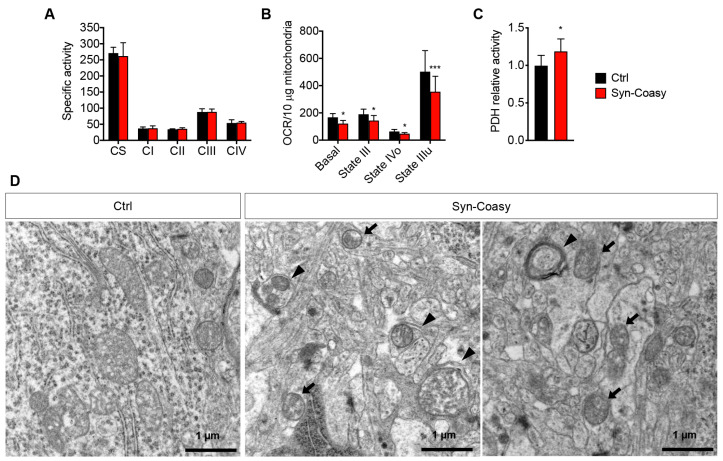
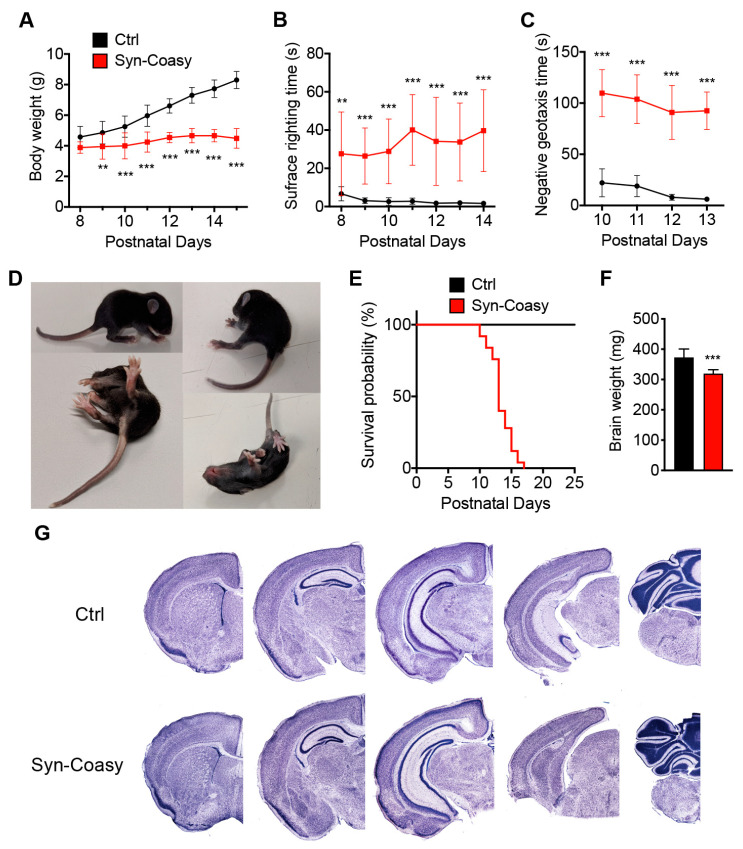
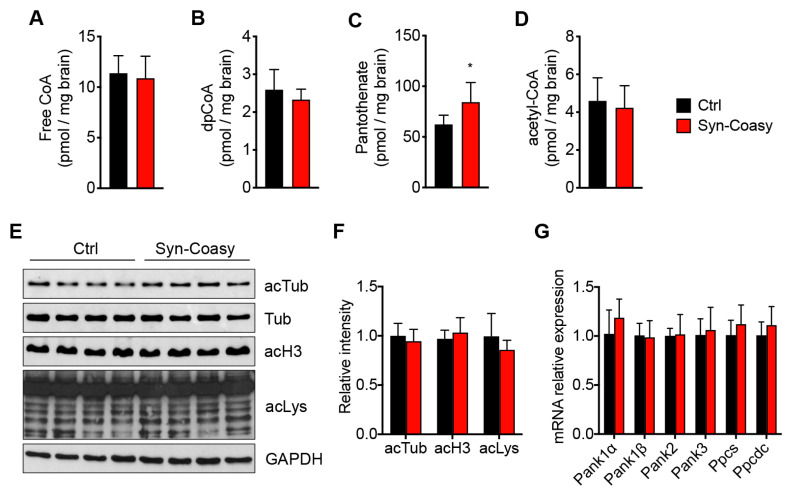
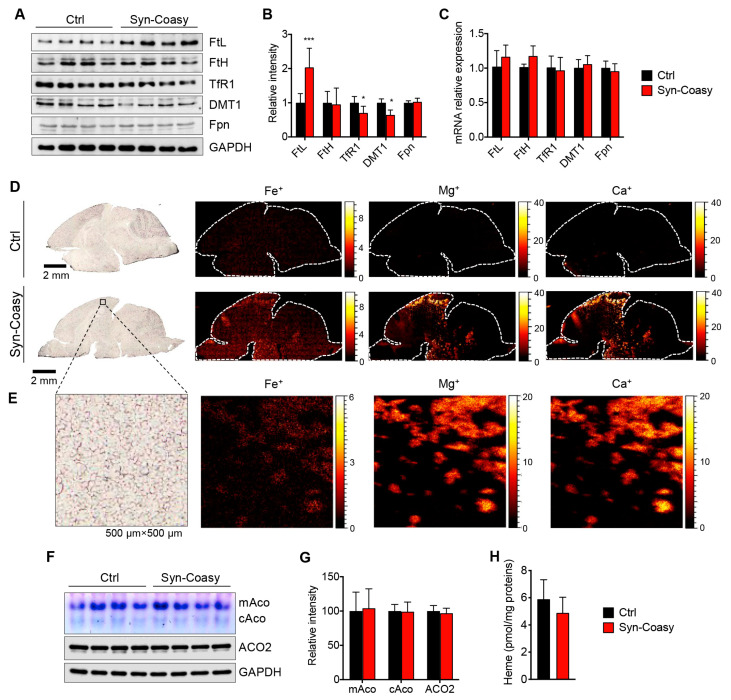
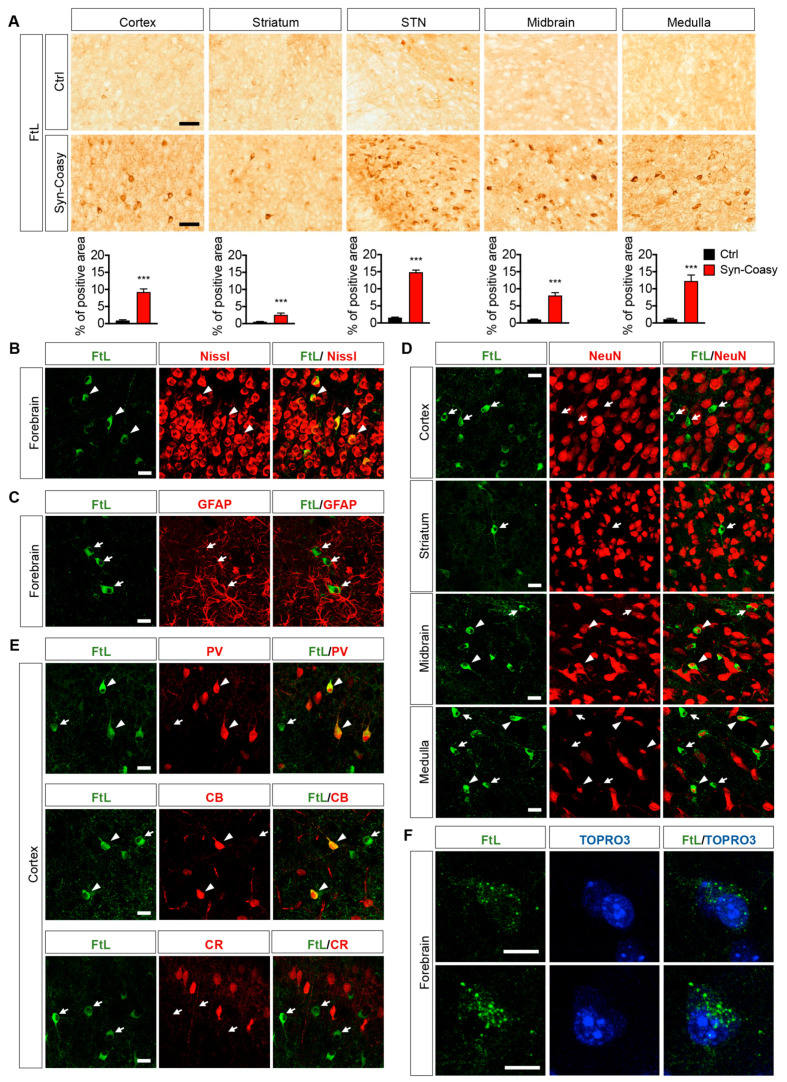
COASY protein-associated neurodegeneration (CoPAN) is a rare but devastating genetic autosomal recessive disorder of inborn error of CoA metabolism, which shares with pantothenate kinase-associated neurodegeneration (PKAN) similar features, such as dystonia, parkinsonian traits, cognitive impairment, axonal neuropathy, and brain iron accumulation. These two disorders are part of the big group of neurodegenerations with brain iron accumulation (NBIA) for which no effective treatment is available at the moment. To date, the lack of a mammalian model, fully recapitulating the human disorder, has prevented the elucidation of pathogenesis and the development of therapeutic approaches. To gain new insights into the mechanisms linking CoA metabolism, iron dyshomeostasis, and neurodegeneration, we generated and characterized the first CoPAN disease mammalian model. Since CoA is a crucial metabolite, constitutive ablation of the gene is incompatible with life. On the contrary, a conditional neuronal-specific knock-out mouse model consistently developed a severe early onset neurological phenotype characterized by sensorimotor defects and dystonia-like movements, leading to premature death. For the first time, we highlighted defective brain iron homeostasis, elevation of iron, calcium, and magnesium, together with mitochondrial dysfunction. Surprisingly, total brain CoA levels were unchanged, and no signs of neurodegeneration were present.
辅酶A合成酶相关神经变性病(CoPAN)是一种罕见但具有毁灭性的常染色体隐性遗传疾病,属于先天性辅酶A代谢缺陷病,与泛酸激酶相关神经变性病(PKAN)具有相似特征,如肌张力障碍、帕金森氏症特征、认知障碍、轴索性神经病和脑铁蓄积。这两种疾病是脑铁蓄积神经变性病(NBIA)大类的一部分,目前尚无有效治疗方法。迄今为止,由于缺乏完全重现人类疾病的哺乳动物模型,阻碍了对发病机制的阐明和治疗方法的开发。为了深入了解辅酶A代谢、铁稳态失衡和神经变性之间的联系机制,我们构建并鉴定了首个CoPAN疾病哺乳动物模型。由于辅酶A是一种关键代谢物,该基因的组成性缺失与生命不相容。相反,条件性神经元特异性基因敲除小鼠模型持续出现严重的早发性神经表型,其特征为感觉运动缺陷和肌张力障碍样运动,导致过早死亡。我们首次发现脑铁稳态失衡、铁、钙和镁升高以及线粒体功能障碍。令人惊讶的是,全脑辅酶A水平未发生变化,且未出现神经变性迹象。