Tang Lu, Chen Yuqiao, Peng Xiong, Zhou Yuan, Jiang Hong, Wang Guo, Zhuang Wei
Department of Thoracic Surgery, Xiangya Hospital, Central South University, Changsha, China.
Department of Thoracic Surgery, The Second Xiangya Hospital, Central South University, Changsha, China.
Front Genet. 2020 Dec 10;11:521004. doi: 10.3389/fgene.2020.521004. eCollection 2020.
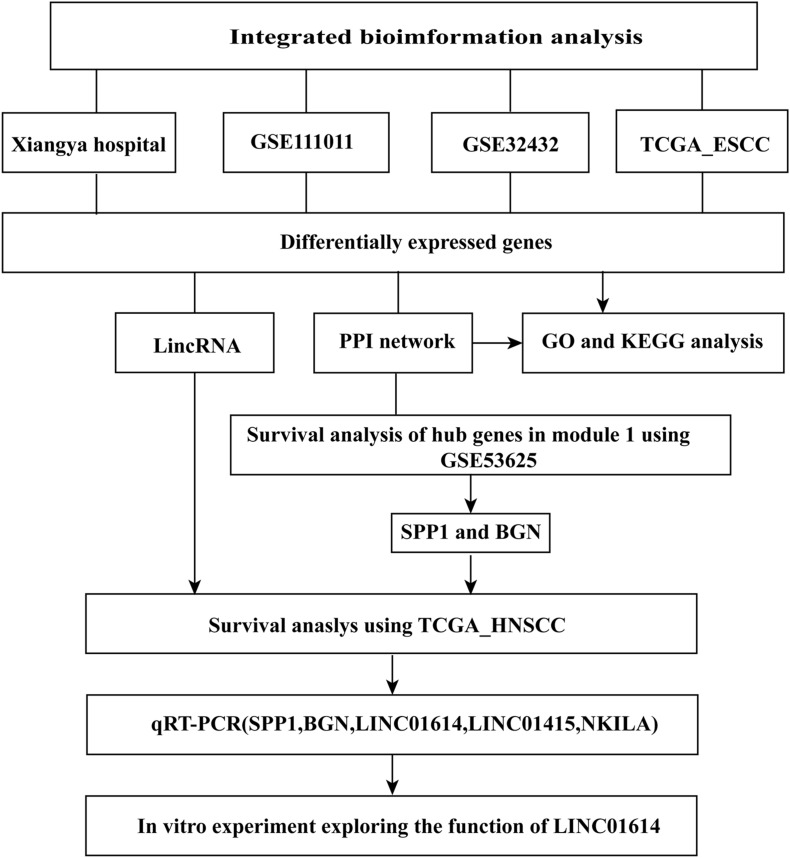
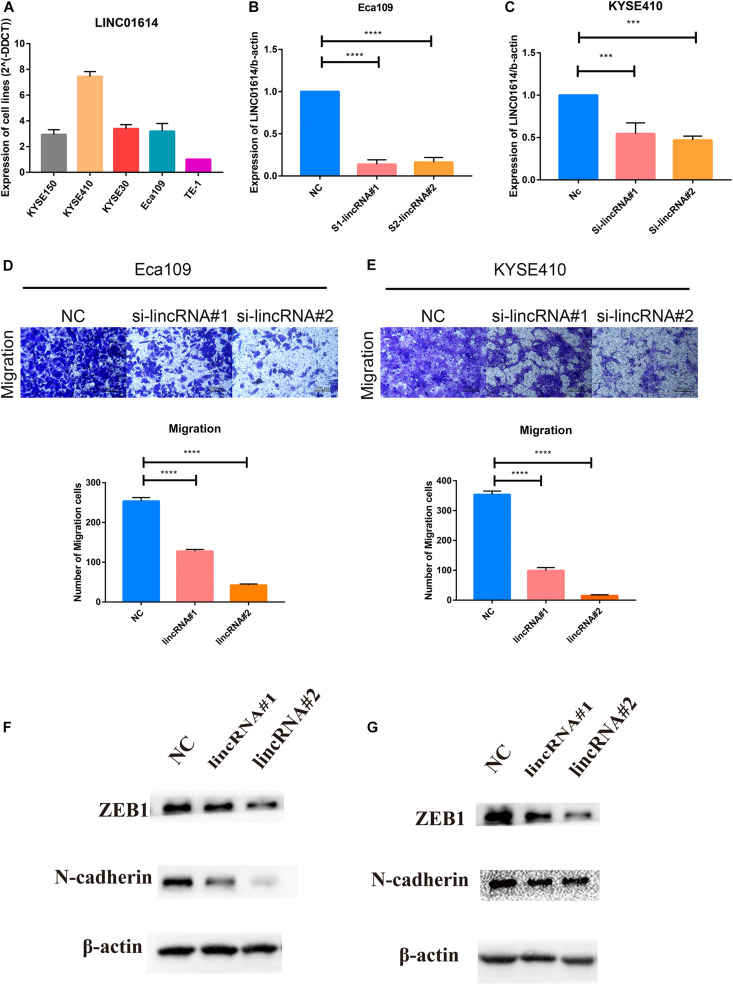
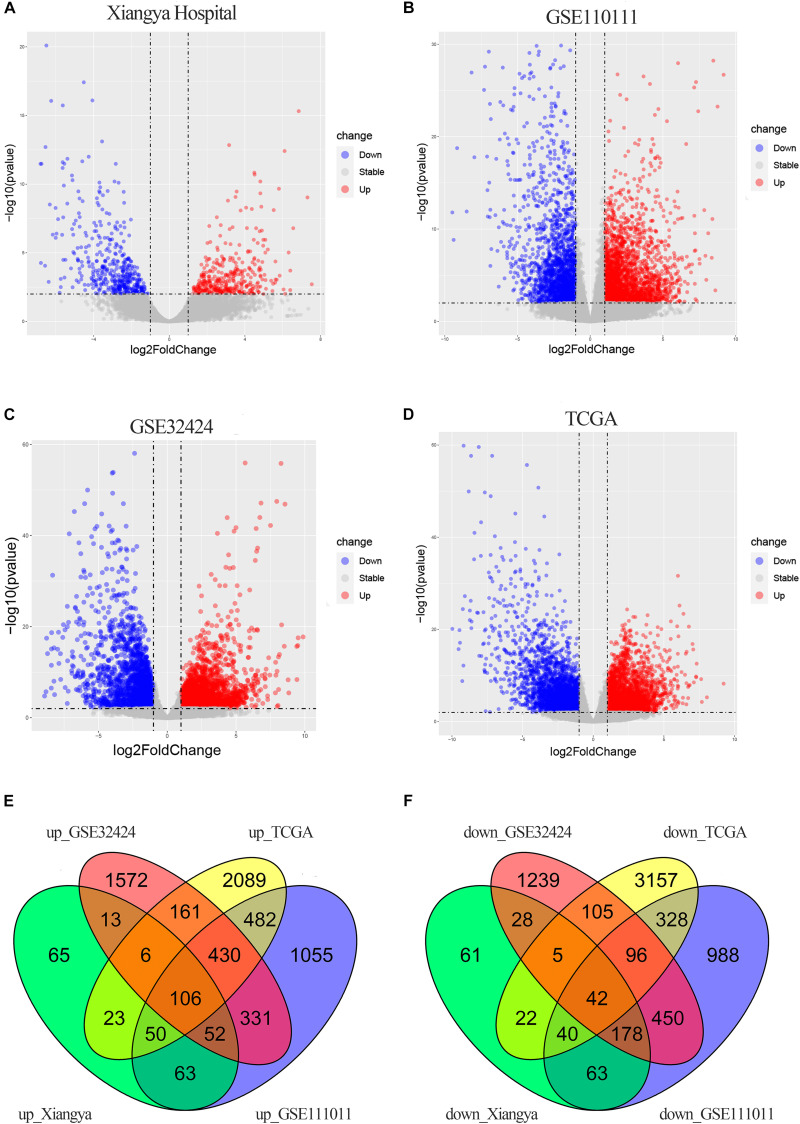
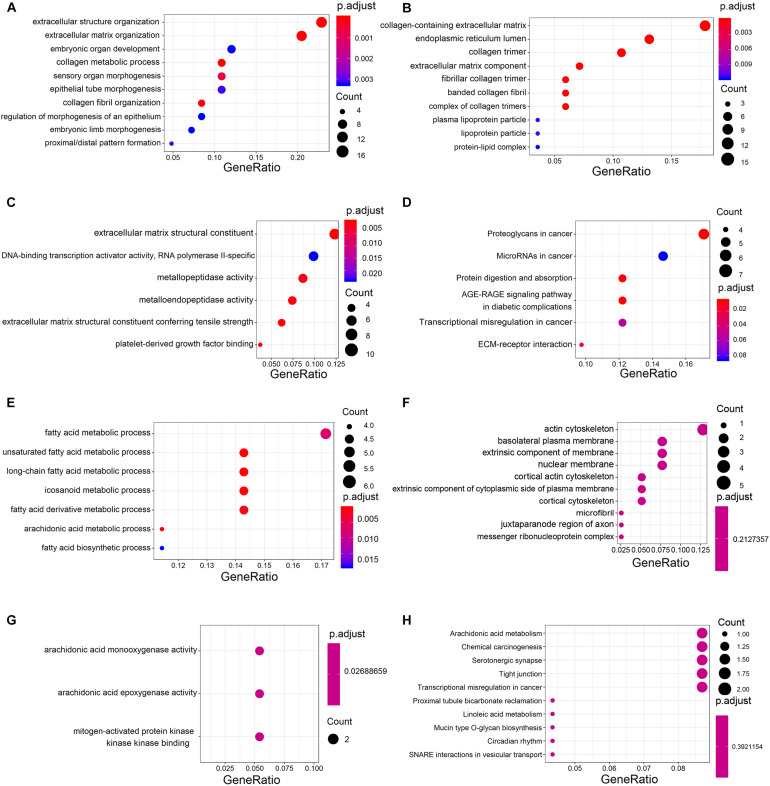
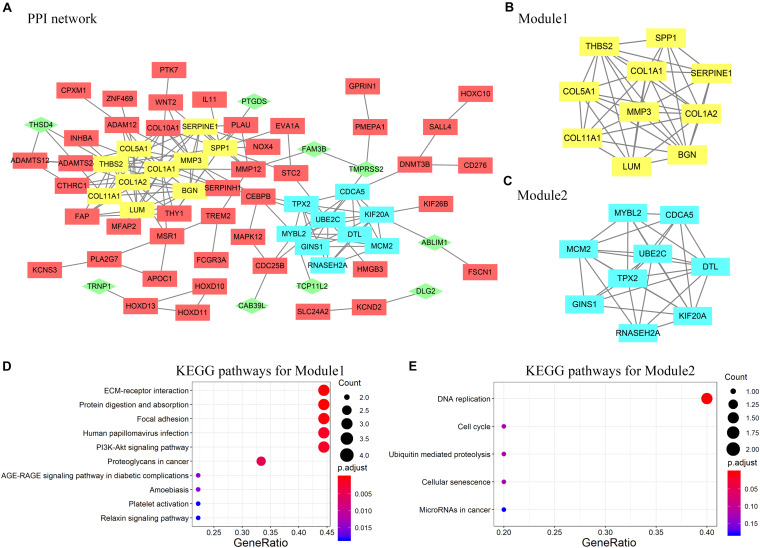
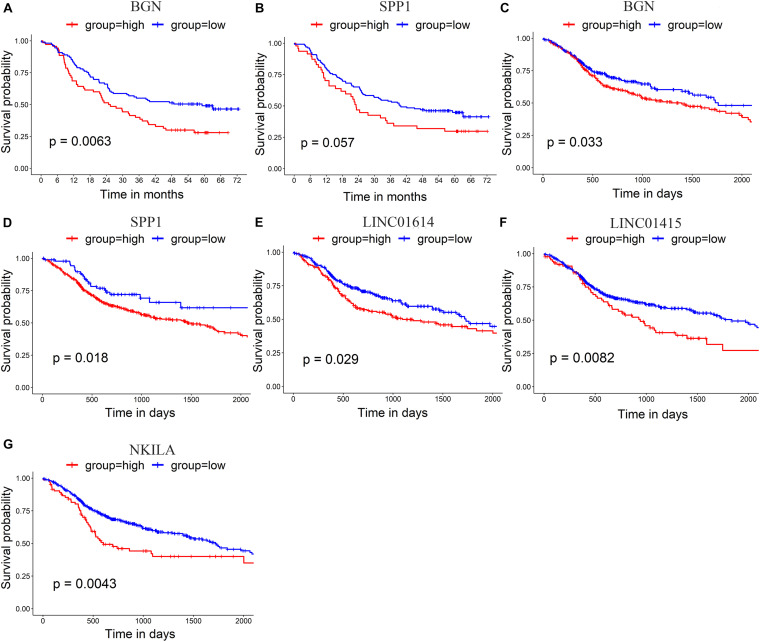
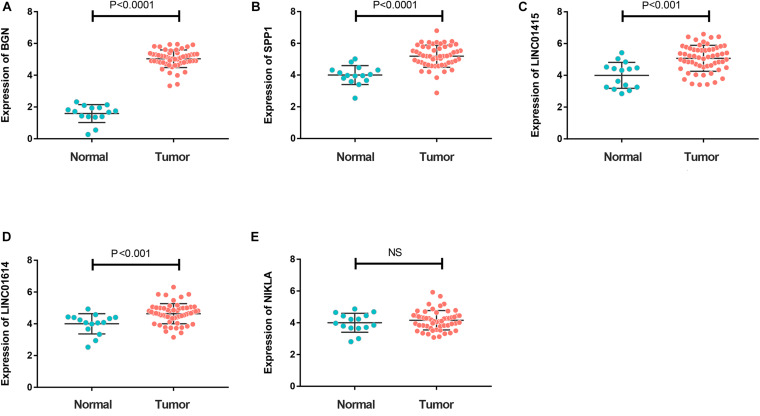
Esophageal squamous cell carcinoma (ESCC) is one of the most fatal malignancies of the digestive tract, but its underlying molecular mechanisms are not known. We aim to identify the genes involved in ESCC carcinogenesis and discover potential prognostic markers using integrated bioinformatics analysis. Three pairs of ESCC tissues and paired normal tissues were sequenced by high-throughput RNA sequencing (RNA-seq). Integrated bioinformatics analysis was used to identify differentially expressed coding genes (DECGs) and differentially expressed long non-coding RNA (lncRNA) genes (DELGs). A protein-protein interaction (PPI) network of DECGs was established using the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) website and visualized with Cytoscape. Survival analysis was conducted by log-rank tests to identify "hub" genes with potential prognostic value, and real-time reverse transcription-quantitative polymerase chain reaction (RT-qPCR) was conducted to assess expression of these genes in ESCC tissues. Transwell assays were employed to examine the migration ability of cells after knockdown of expression, followed by investigation of epithelial-mesenchymal transition (EMT) by western blotting (WB). A total of 106 upregulated genes and 42 downregulated genes were screened out from the ESCC data sets. Survival analysis showed two hub protein-coding genes with higher expression in module 1 of the PPI network ( and ) and another three upregulated lncRNAs (, , ) that were associated with a poor prognosis. High expression of , , , and in tumor samples was validated further by RT-qPCR. experiments show that knockdown of expression could significantly inhibit the migration of ESCC cells by regulating EMT, which was confirmed by WB. These results indicate that , , , and might be critical genes in ESCC and potential prognostic biomarkers.
食管鳞状细胞癌(ESCC)是消化道最致命的恶性肿瘤之一,但其潜在的分子机制尚不清楚。我们旨在通过综合生物信息学分析,鉴定参与ESCC致癌过程的基因,并发现潜在的预后标志物。通过高通量RNA测序(RNA-seq)对三对ESCC组织和配对的正常组织进行测序。综合生物信息学分析用于鉴定差异表达的编码基因(DECGs)和差异表达的长链非编码RNA(lncRNA)基因(DELGs)。使用检索相互作用基因/蛋白质的搜索工具(STRING)网站建立DECGs的蛋白质-蛋白质相互作用(PPI)网络,并用Cytoscape进行可视化。通过对数秩检验进行生存分析,以鉴定具有潜在预后价值的“枢纽”基因,并进行实时逆转录定量聚合酶链反应(RT-qPCR)以评估这些基因在ESCC组织中的表达。采用Transwell实验检测基因敲低后细胞的迁移能力,随后通过蛋白质印迹法(WB)研究上皮-间质转化(EMT)。从ESCC数据集中筛选出106个上调基因和42个下调基因。生存分析显示,PPI网络模块1中两个表达较高的枢纽蛋白编码基因(和)以及另外三个上调的lncRNAs(、、)与预后不良相关。RT-qPCR进一步验证了肿瘤样本中、、、的高表达。实验表明,敲低基因表达可通过调节EMT显著抑制ESCC细胞的迁移,WB证实了这一点。这些结果表明,、、、可能是ESCC中的关键基因和潜在的预后生物标志物。