Lei Kuan Cheok, Zhang Xiaohua Douglas
CRDA, Faculty of Health Sciences, University of Macau, Macau, China.
Evol Med Public Health. 2020 Nov 5;2020(1):290-303. doi: 10.1093/emph/eoaa041. eCollection 2020.
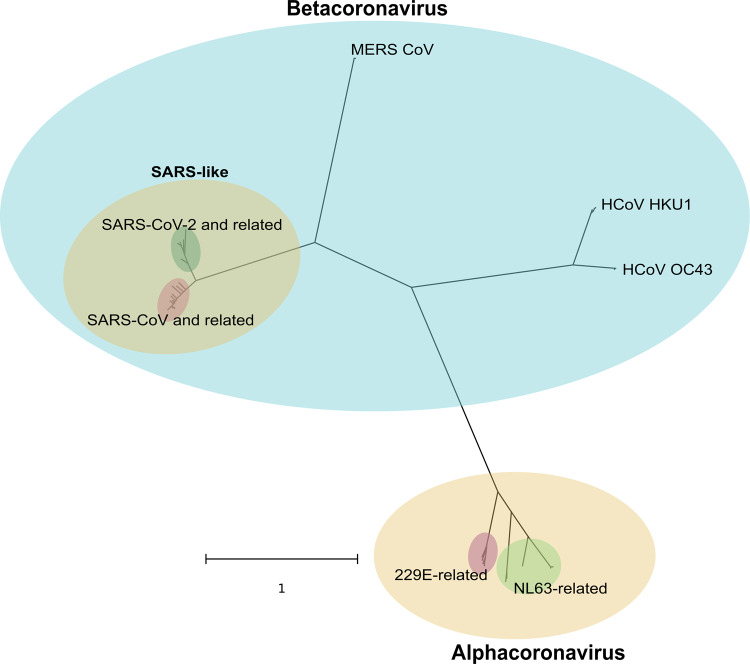
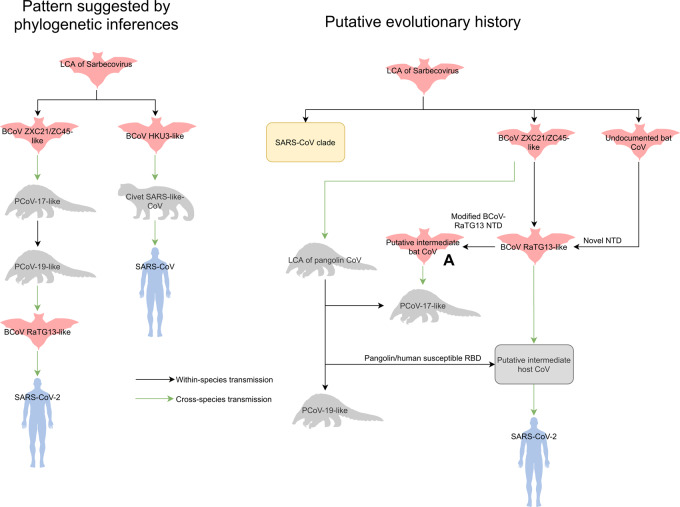
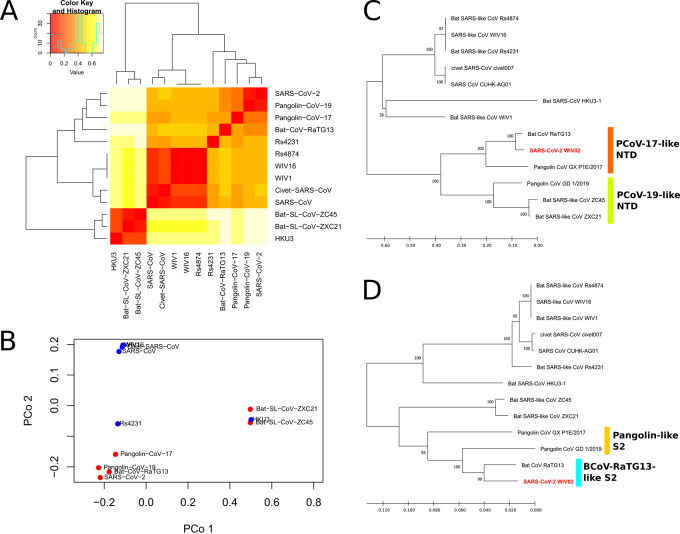
The current coronavirus disease 2019 (COVID-19) pandemic, caused by severe acute respiratory syndrome (SARS)-CoV-2, has become the most devastating public health emergency in the 21st century and one of the most influential plagues in history. Studies on the origin of SARS-CoV-2 have generally agreed that the virus probably comes from bat, closely related to a bat CoV named BCoV-RaTG13 taken from horseshoe bat (), with Malayan pangolin () being a plausible intermediate host. However, due to the relatively low number of SARS-CoV-2-related strains available in public domain, the evolutionary history remains unclear.
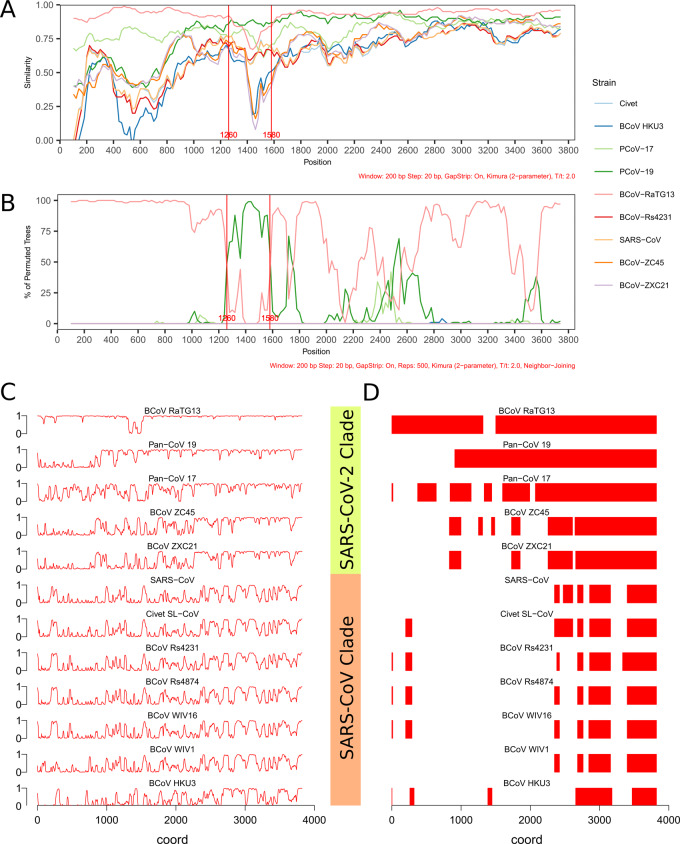
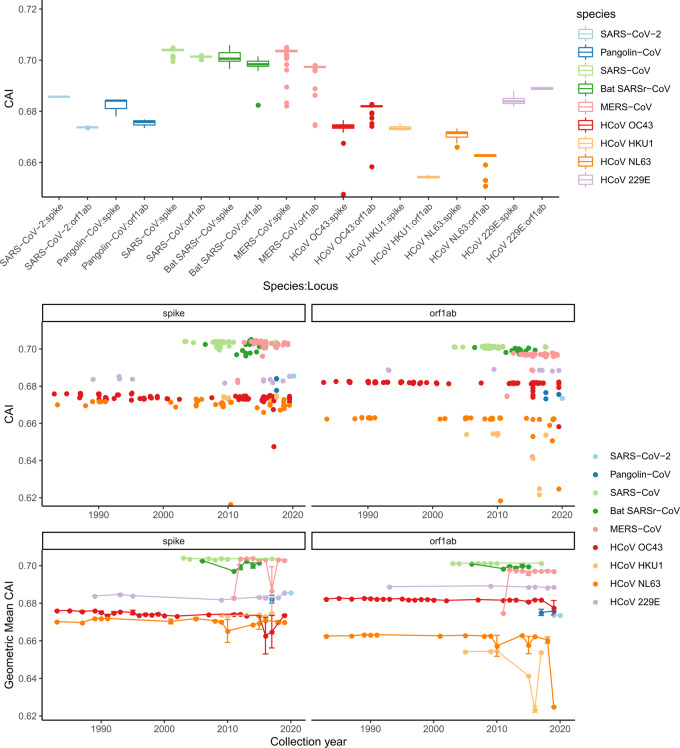
Nine hundred ninety-five coronavirus sequences from NCBI Genbank and GISAID were obtained and multiple sequence alignment was carried out to categorize SARS-CoV-2 related groups. Spike sequences were analyzed using similarity analysis and conservation analyses. Mutation analysis was used to identify variations within receptor-binding domain (RBD) in spike for SARS-CoV-2-related strains.
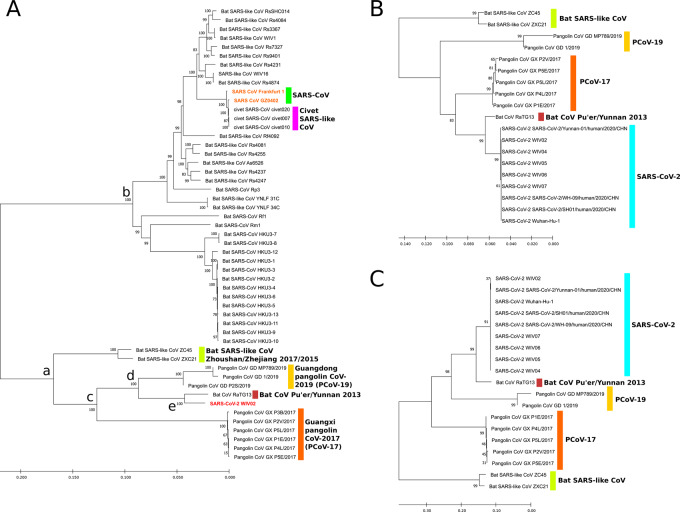
We identified a family of SARS-CoV-2-related strains, including the closest relatives, bat CoV RaTG13 and pangolin CoV strains. Sequence similarity analysis and conservation analysis on spike sequence identified that N-terminal domain, RBD and S2 subunit display different degrees of conservation with several coronavirus strains. Mutation analysis on contact sites in SARS-CoV-2 RBD reveals that human-susceptibility probably emerges in pangolin.
We conclude that the spike sequence of SARS-CoV-2 is the result of multiple recombination events during its transmission from bat to human, and we propose a framework of evolutionary history that resolve the relationship of BCoV-RaTG13 and pangolin coronaviruses with SARS-CoV-2.
This study analyses whole-genome and spike sequences of coronavirus from NCBI using phylogenetic and conservation analyses to reconstruct the evolutionary history of severe acute respiratory syndrome (SARS)-CoV-2 and proposes an evolutionary history of spike in the progenitors of SARS-CoV-2 from bat to human through mammal hosts before they recombine into the current form.
由严重急性呼吸综合征冠状病毒 2 型(SARS-CoV-2)引发的 2019 冠状病毒病(COVID-19)大流行已成为 21 世纪最具毁灭性的突发公共卫生事件,也是历史上最具影响力的瘟疫之一。关于 SARS-CoV-2 起源的研究普遍认为,该病毒可能源自蝙蝠,与从菊头蝠体内提取的一株名为 BCoV-RaTG13 的蝙蝠冠状病毒密切相关,马来穿山甲可能是一个合理的中间宿主。然而,由于公共数据库中与 SARS-CoV-2 相关的毒株数量相对较少,其进化史仍不明确。
从 NCBI 基因库和 GISAID 获取了 995 条冠状病毒序列,并进行了多序列比对以对与 SARS-CoV-2 相关的群体进行分类。使用相似性分析和保守性分析对刺突序列进行了分析。突变分析用于识别 SARS-CoV-2 相关毒株刺突蛋白受体结合域(RBD)内的变异。
我们鉴定出了一个与 SARS-CoV-2 相关的毒株家族,包括其近亲蝙蝠冠状病毒 RaTG13 和穿山甲冠状病毒毒株。对刺突序列的相似性分析和保守性分析表明,N 端结构域、RBD 和 S2 亚基与几种冠状病毒毒株呈现出不同程度的保守性。对 SARS-CoV-2 RBD 接触位点的突变分析表明,人易感性可能在穿山甲中出现。
我们得出结论,SARS-CoV-2 的刺突序列是其从蝙蝠传播至人类过程中多次重组事件的结果,并且我们提出了一个进化史框架,该框架解析了 BCoV-RaTG13 和穿山甲冠状病毒与 SARS-CoV-2 之间的关系。
本研究使用系统发育分析和保守性分析对来自 NCBI 的冠状病毒全基因组和刺突序列进行分析,以重建严重急性呼吸综合征冠状病毒 2 型(SARS-CoV-2)的进化史,并提出了 SARS-CoV-2 祖先从蝙蝠通过哺乳动物宿主传播至人类并重组为当前形式之前刺突蛋白的进化史。