Pace Josh, Youens-Clark Ken, Freeman Cordell, Hurwitz Bonnie, Van Doorslaer Koenraad
School of Animal and Comparative Biomedical Sciences, University of Arizona, 1200 E. University Blvd. Tucson, AZ 85721-0073, USA.
Department of Biosystems Engineering, University of Arizona, 1200 E. University Blvd. Tucson, AZ 85721-0073, USA.
Virus Evol. 2020 Aug 26;6(2):veaa068. doi: 10.1093/ve/veaa068. eCollection 2020 Jul.
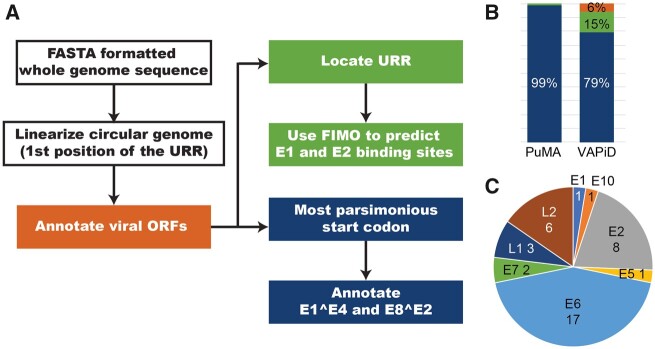
High-throughput sequencing technologies provide unprecedented power to identify novel viruses from a wide variety of (environmental) samples. The field of 'viral metagenomics' has dramatically expanded our understanding of viral diversity. Viral metagenomic approaches imply that many novel viruses will not be described by researchers who are experts on (the genomic organization of) that virus family. We have developed the papillomavirus annotation tool (PuMA) to provide researchers with a convenient and reproducible method to annotate and report novel papillomaviruses. PuMA currently correctly annotates 99% of the papillomavirus genes when benchmarked against the 655 reference genomes in the papillomavirus episteme. Compared to another viral annotation pipeline, PuMA annotates more viral features while being more accurate. To demonstrate its general applicability, we also developed a preliminary version of PuMA that can annotate polyomaviruses. PuMA is available on GitHub (https://github.com/KVD-lab/puma) and through the iMicrobe online environment (https://www.imicrobe.us/#/apps/puma).
高通量测序技术为从各种(环境)样本中鉴定新型病毒提供了前所未有的能力。“病毒宏基因组学”领域极大地扩展了我们对病毒多样性的理解。病毒宏基因组学方法意味着许多新型病毒不会被该病毒家族(基因组组织)方面的专家研究人员所描述。我们开发了乳头瘤病毒注释工具(PuMA),为研究人员提供一种方便且可重复的方法来注释和报告新型乳头瘤病毒。当以乳头瘤病毒知识体系中的655个参考基因组为基准进行测试时,PuMA目前能正确注释99%的乳头瘤病毒基因。与另一种病毒注释流程相比,PuMA能注释更多病毒特征且更准确。为证明其普遍适用性,我们还开发了一个能注释多瘤病毒的PuMA初步版本。PuMA可在GitHub(https://github.com/KVD-lab/puma)以及通过iMicrobe在线环境(https://www.imicrobe.us/#/apps/puma)获取。