Departments of Neurosciences, Experimental Neurology, and Leuven Brain Institute (LBI), KU Leuven - University of Leuven, Leuven, Belgium.
Laboratory of Neurobiology, VIB, Center for Brain and Disease Research, Leuven, Belgium.
Ann Neurol. 2021 Apr;89(4):686-697. doi: 10.1002/ana.26009. Epub 2021 Jan 15.
The role of the survival of motor neuron (SMN) gene in amyotrophic lateral sclerosis (ALS) is unclear, with several conflicting reports. A decisive result on this topic is needed, given that treatment options are available now for SMN deficiency.
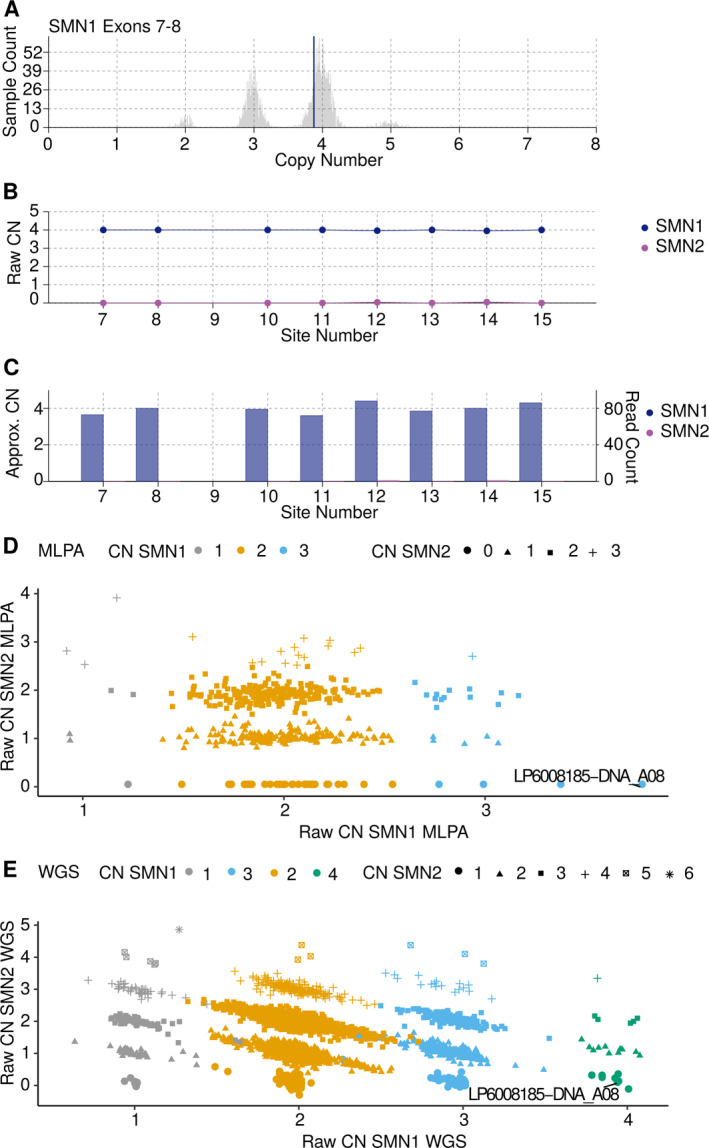
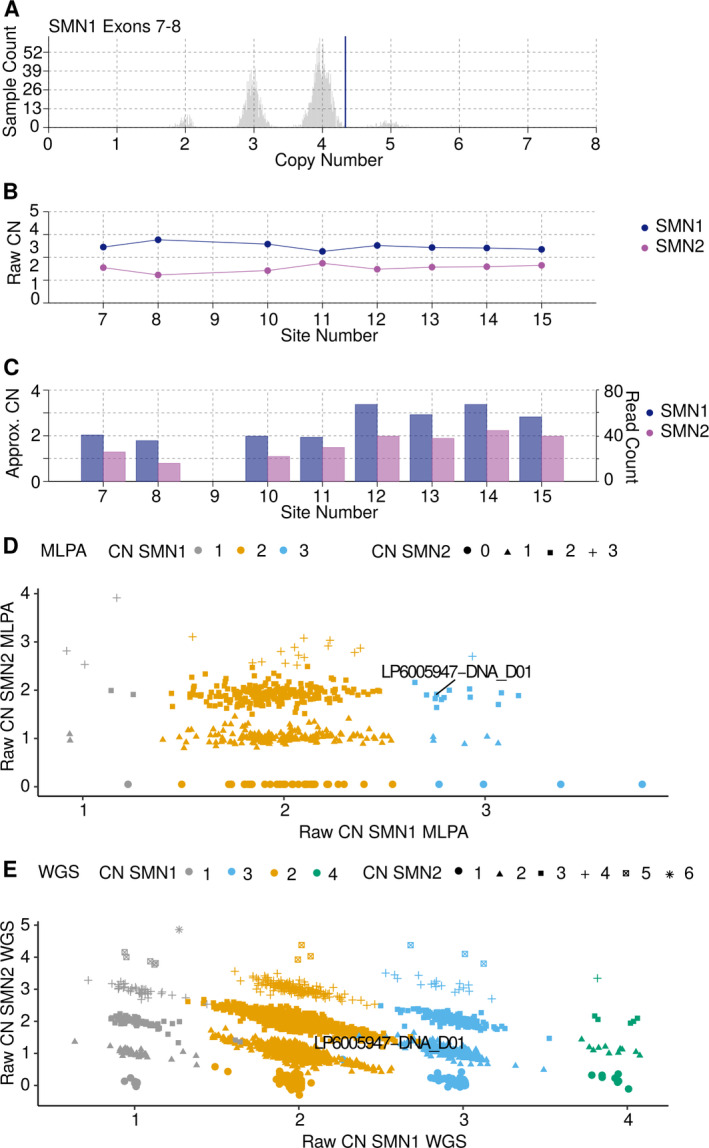
In this largest multicenter case control study to evaluate the effect of SMN1 and SMN2 copy numbers in ALS, we used whole genome sequencing data from Project MinE data freeze 2. SMN copy numbers of 6,375 patients with ALS and 2,412 controls were called from whole genome sequencing data, and the reliability of the calls was tested with multiplex ligation-dependent probe amplification data.
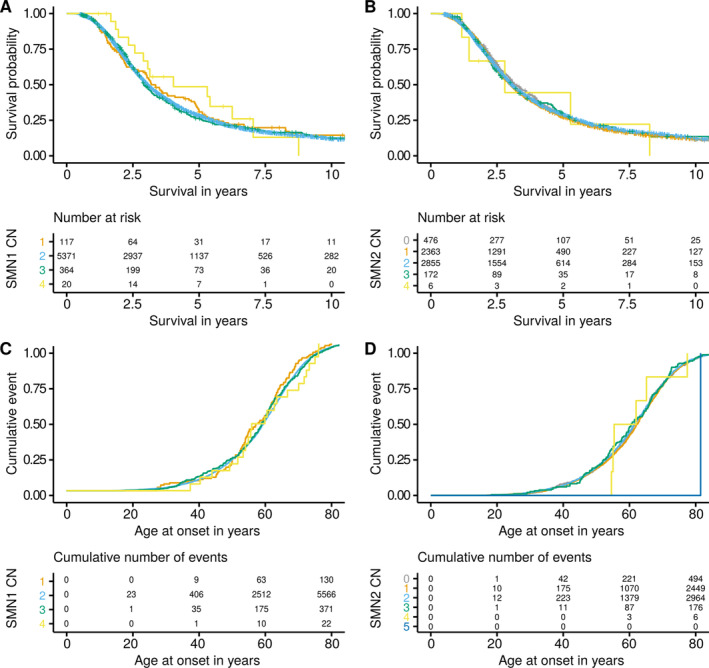
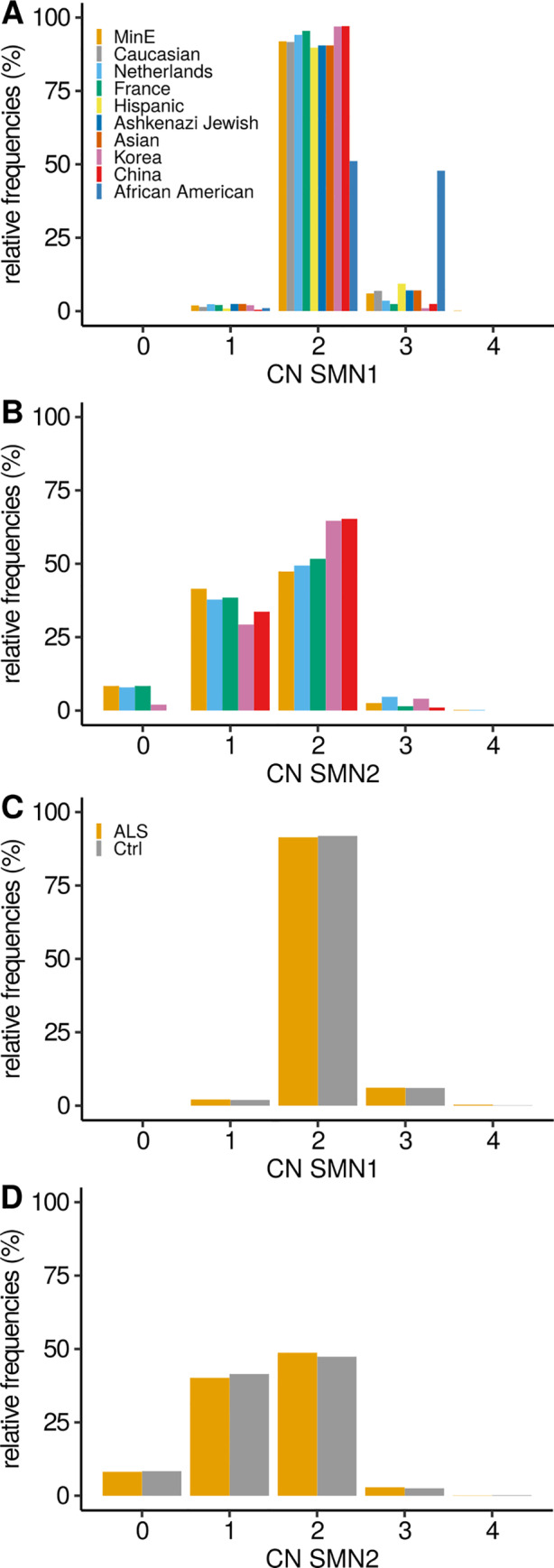
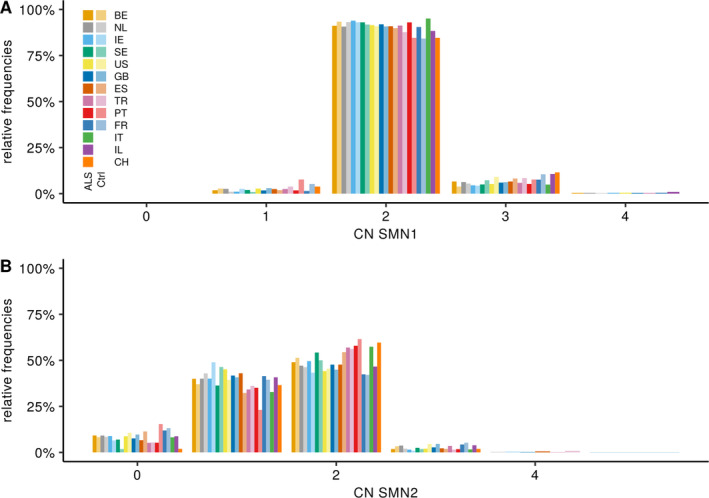
The copy number distribution of SMN1 and SMN2 between cases and controls did not show any statistical differences (binomial multivariate logistic regression SMN1 p = 0.54 and SMN2 p = 0.49). In addition, the copy number of SMN did not associate with patient survival (Royston-Parmar; SMN1 p = 0.78 and SMN2 p = 0.23) or age at onset (Royston-Parmar; SMN1 p = 0.75 and SMN2 p = 0.63).
In our well-powered study, there was no association of SMN1 or SMN2 copy numbers with the risk of ALS or ALS disease severity. This suggests that changing SMN protein levels in the physiological range may not modify ALS disease course. This is an important finding in the light of emerging therapies targeted at SMN deficiencies. ANN NEUROL 2021;89:686-697.
运动神经元存活(SMN)基因在肌萎缩侧索硬化症(ALS)中的作用尚不清楚,有几项相互矛盾的报告。鉴于现在有针对 SMN 缺乏的治疗选择,需要对此主题进行决定性的研究结果。
在这项评估 SMN1 和 SMN2 拷贝数在 ALS 中作用的最大多中心病例对照研究中,我们使用了 Project MinE 数据冻结 2 中的全基因组测序数据。从全基因组测序数据中调用了 6375 例 ALS 患者和 2412 例对照者的 SMN 拷贝数,并使用多重连接依赖性探针扩增数据测试了这些调用的可靠性。
病例和对照者之间的 SMN1 和 SMN2 的拷贝数分布没有显示出任何统计学差异(二项式多元逻辑回归 SMN1 p = 0.54 和 SMN2 p = 0.49)。此外,SMN 的拷贝数与患者生存(Royston-Parmar;SMN1 p = 0.78 和 SMN2 p = 0.23)或发病年龄(Royston-Parmar;SMN1 p = 0.75 和 SMN2 p = 0.63)无关。
在我们这项效能良好的研究中,SMN1 或 SMN2 拷贝数与 ALS 的发病风险或 ALS 疾病严重程度均无关联。这表明,在生理范围内改变 SMN 蛋白水平可能不会改变 ALS 的病程。鉴于针对 SMN 缺乏的新兴疗法,这是一个重要的发现。神经病学年鉴 2021;89:686-697。