Shenzhen Neocura Biotechnology Co. Ltd., Shenzhen, 518055, China.
School of Computer Science and Technology, Heilongjiang University, Harbin, 150080, China.
BMC Bioinformatics. 2021 Jan 6;22(1):7. doi: 10.1186/s12859-020-03946-z.
Accurate prediction of binding between class I human leukocyte antigen (HLA) and neoepitope is critical for target identification within personalized T-cell based immunotherapy. Many recent prediction tools developed upon the deep learning algorithms and mass spectrometry data have indeed showed improvement on the average predicting power for class I HLA-peptide interaction. However, their prediction performances show great variability over individual HLA alleles and peptides with different lengths, which is particularly the case for HLA-C alleles due to the limited amount of experimental data. To meet the increasing demand for attaining the most accurate HLA-peptide binding prediction for individual patient in the real-world clinical studies, more advanced deep learning framework with higher prediction accuracy for HLA-C alleles and longer peptides is highly desirable.
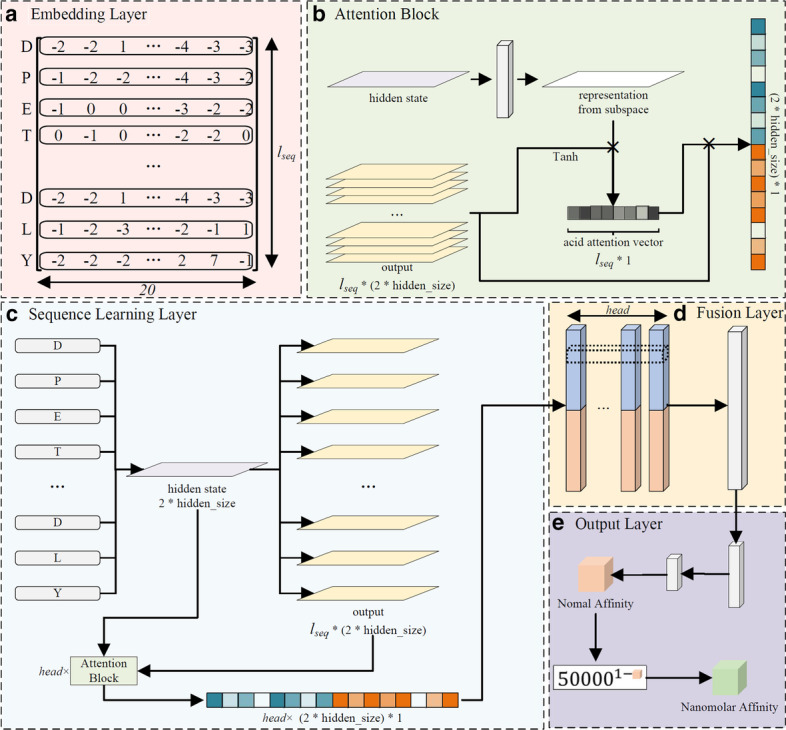
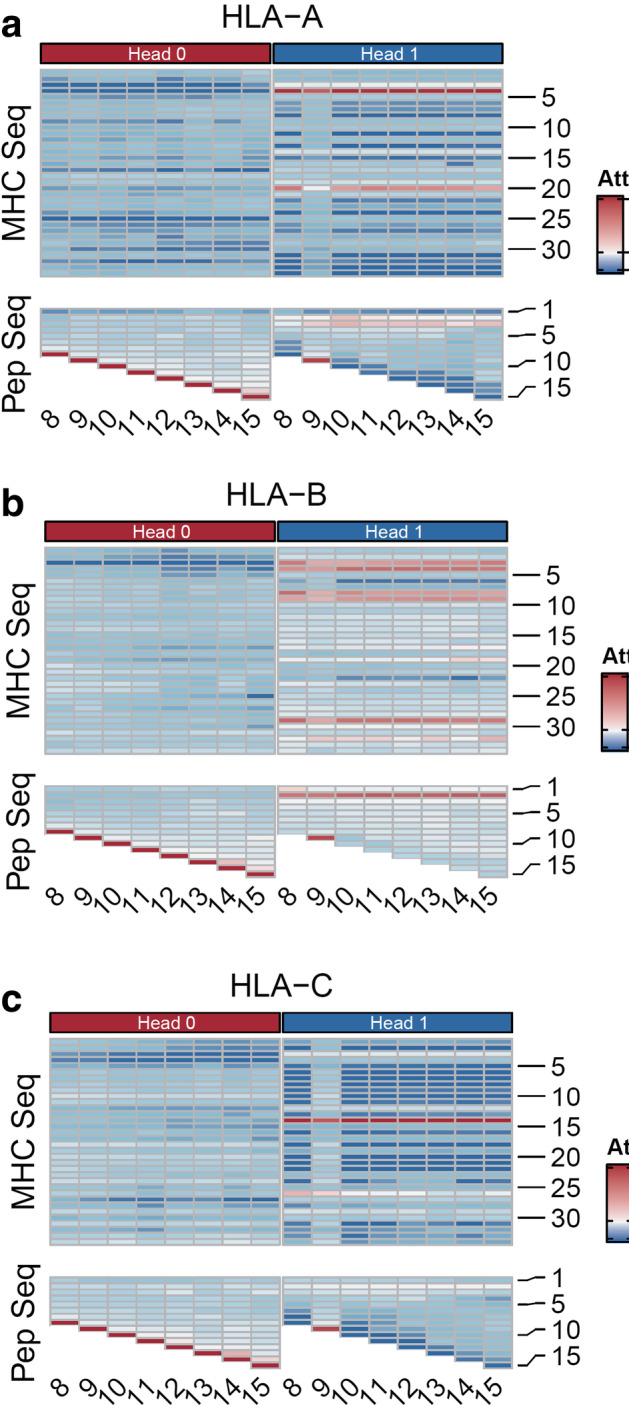
We present a pan-allele HLA-peptide binding prediction framework-MATHLA which integrates bi-directional long short-term memory network and multiple head attention mechanism. This model achieves better prediction accuracy in both fivefold cross-validation test and independent test dataset. In addition, this model is superior over existing tools regarding to the prediction accuracy for longer ligand ranging from 11 to 15 amino acids. Moreover, our model also shows a significant improvement for HLA-C-peptide-binding prediction. By investigating multiple-head attention weight scores, we depicted possible interaction patterns between three HLA I supergroups and their cognate peptides.
Our method demonstrates the necessity of further development of deep learning algorithm in improving and interpreting HLA-peptide binding prediction in parallel to increasing the amount of high-quality HLA ligandome data.
准确预测 I 类人类白细胞抗原(HLA)与新表位之间的结合对于个性化 T 细胞免疫治疗中的靶标识别至关重要。许多最近基于深度学习算法和质谱数据开发的预测工具确实提高了 I 类 HLA-肽相互作用的平均预测能力。然而,它们的预测性能在个体 HLA 等位基因和不同长度的肽之间表现出很大的可变性,对于 HLA-C 等位基因尤其如此,因为实验数据有限。为了满足在真实世界临床研究中为个体患者获得最准确 HLA-肽结合预测的需求,需要更先进的深度学习框架,以提高 HLA-C 等位基因和更长肽的预测准确性。
我们提出了一个泛等位基因 HLA-肽结合预测框架-MATHLA,该框架集成了双向长短期记忆网络和多头注意力机制。该模型在五重交叉验证测试和独立测试数据集上都实现了更好的预测准确性。此外,与现有的工具相比,该模型在预测长度为 11 到 15 个氨基酸的更长配体方面具有更高的准确性。此外,我们的模型在 HLA-C-肽结合预测方面也有显著的改进。通过研究多头注意力权重分数,我们描绘了三个 HLA I 超组与其同源肽之间可能的相互作用模式。
我们的方法证明了在增加高质量 HLA 配体组数据的同时,进一步开发深度学习算法对于改进和解释 HLA-肽结合预测的必要性。