Zhou Ji-Ren, You Zhu-Hong, Cheng Li, Ji Bo-Ya
The Xinjiang Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Urumqi 830011, China.
University of Chinese Academy of Sciences, Beijing 100049, China.
Mol Ther Nucleic Acids. 2020 Nov 4;23:277-285. doi: 10.1016/j.omtn.2020.10.040. eCollection 2021 Mar 5.
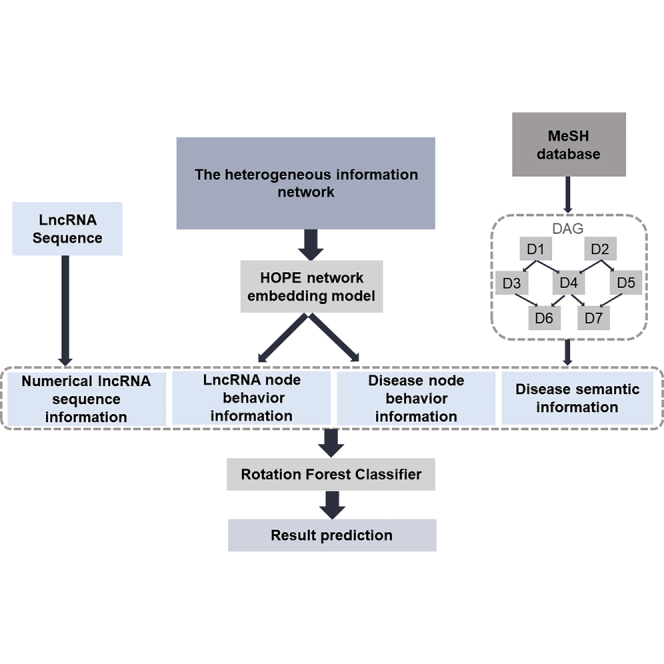
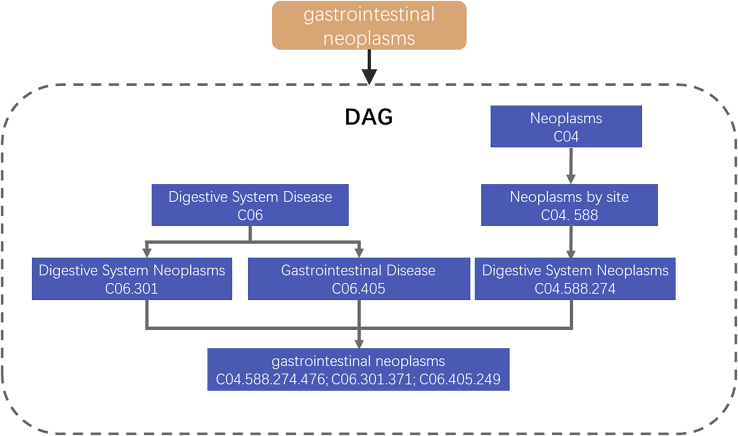
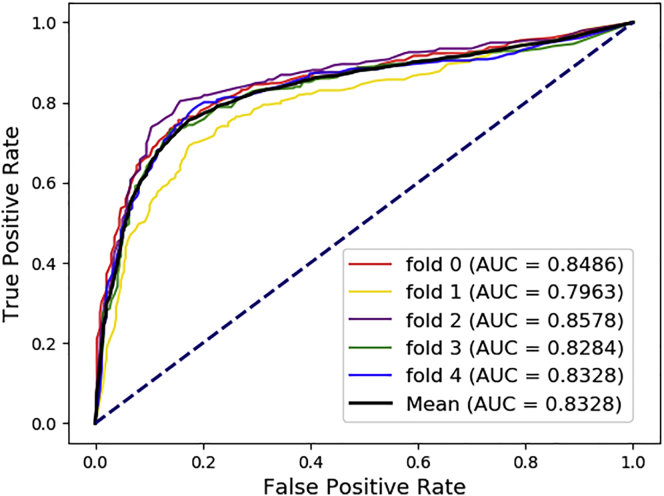
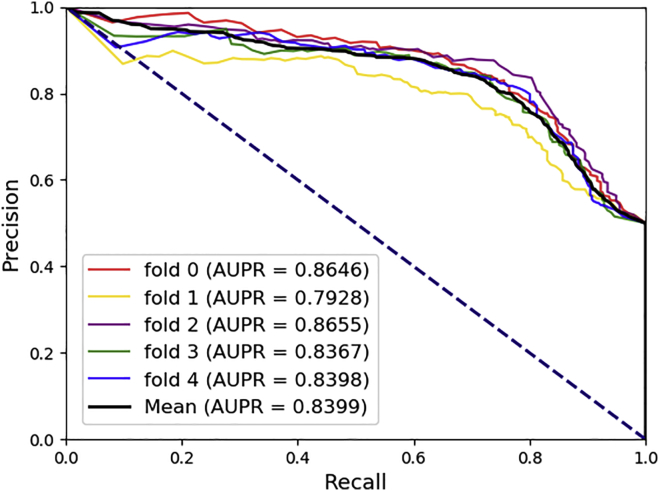
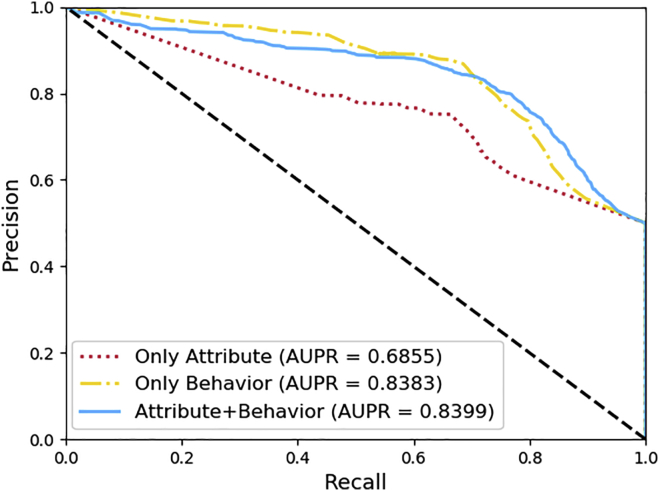
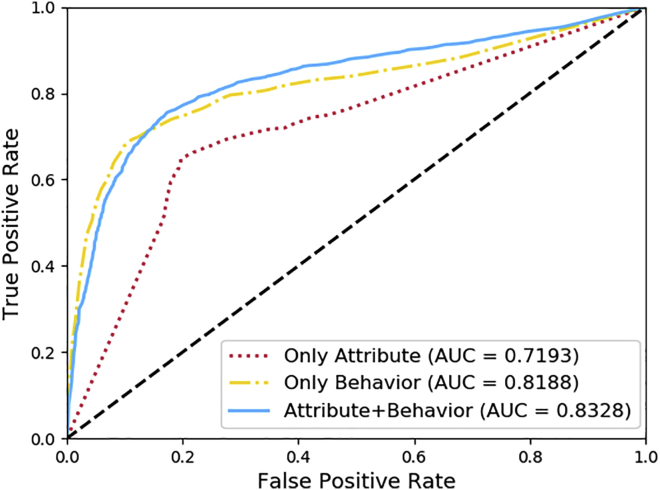
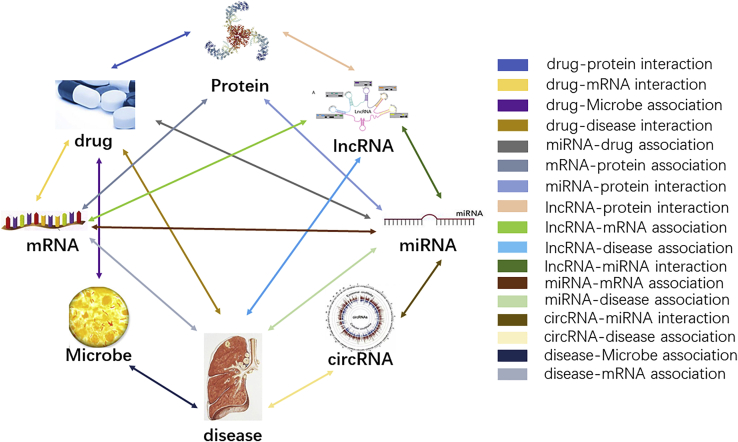
Uncovering additional long non-coding RNA (lncRNA)-disease associations has become increasingly important for developing treatments for complex human diseases. Identification of lncRNA biomarkers and lncRNA-disease associations is central to diagnoses and treatment. However, traditional experimental methods are expensive and time-consuming. Enormous amounts of data present in public biological databases are available for computational methods used to predict lncRNA-disease associations. In this study, we propose a novel computational method to predict lncRNA-disease associations. More specifically, a heterogeneous network is first constructed by integrating the associations among microRNA (miRNA), lncRNA, protein, drug, and disease, Second, high-order proximity preserved embedding (HOPE) was used to embed nodes into a network. Finally, the rotation forest classifier was adopted to train the prediction model. In the 5-fold cross-validation experiment, the area under the curve (AUC) of our method achieved 0.8328 ± 0.0236. We compare it with the other four classifiers, in which the proposed method remarkably outperformed other comparison methods. Otherwise, we constructed three case studies for three excess death rate cancers, respectively. The results show that 9 (lung cancer, gastric cancer, and hepatocellular carcinomas) out of the top 15 predicted disease-related lncRNAs were confirmed by our method. In conclusion, our method could predict the unknown lncRNA-disease associations effectively.
发现更多长链非编码RNA(lncRNA)与疾病的关联对于开发复杂人类疾病的治疗方法变得越来越重要。lncRNA生物标志物和lncRNA与疾病关联的识别是诊断和治疗的核心。然而,传统的实验方法既昂贵又耗时。公共生物数据库中存在大量数据,可用于预测lncRNA与疾病关联的计算方法。在本研究中,我们提出了一种预测lncRNA与疾病关联的新计算方法。更具体地说,首先通过整合微小RNA(miRNA)、lncRNA、蛋白质、药物和疾病之间的关联构建一个异质网络,其次,使用高阶接近度保留嵌入(HOPE)将节点嵌入网络。最后,采用旋转森林分类器训练预测模型。在5折交叉验证实验中,我们方法的曲线下面积(AUC)达到0.8328±0.0236。我们将其与其他四个分类器进行比较,结果表明所提出的方法明显优于其他比较方法。此外,我们分别针对三种超额死亡率癌症构建了三个案例研究。结果表明,我们的方法证实了前15个预测的疾病相关lncRNA中的9个(肺癌、胃癌和肝细胞癌)。总之,我们的方法可以有效地预测未知的lncRNA与疾病的关联。