Friedrich Miescher Institute for Biomedical Research, Basel, Switzerland.
SIB Swiss Institute of Bioinformatics, Basel, Switzerland.
PLoS Comput Biol. 2021 Jan 11;17(1):e1008585. doi: 10.1371/journal.pcbi.1008585. eCollection 2021 Jan.
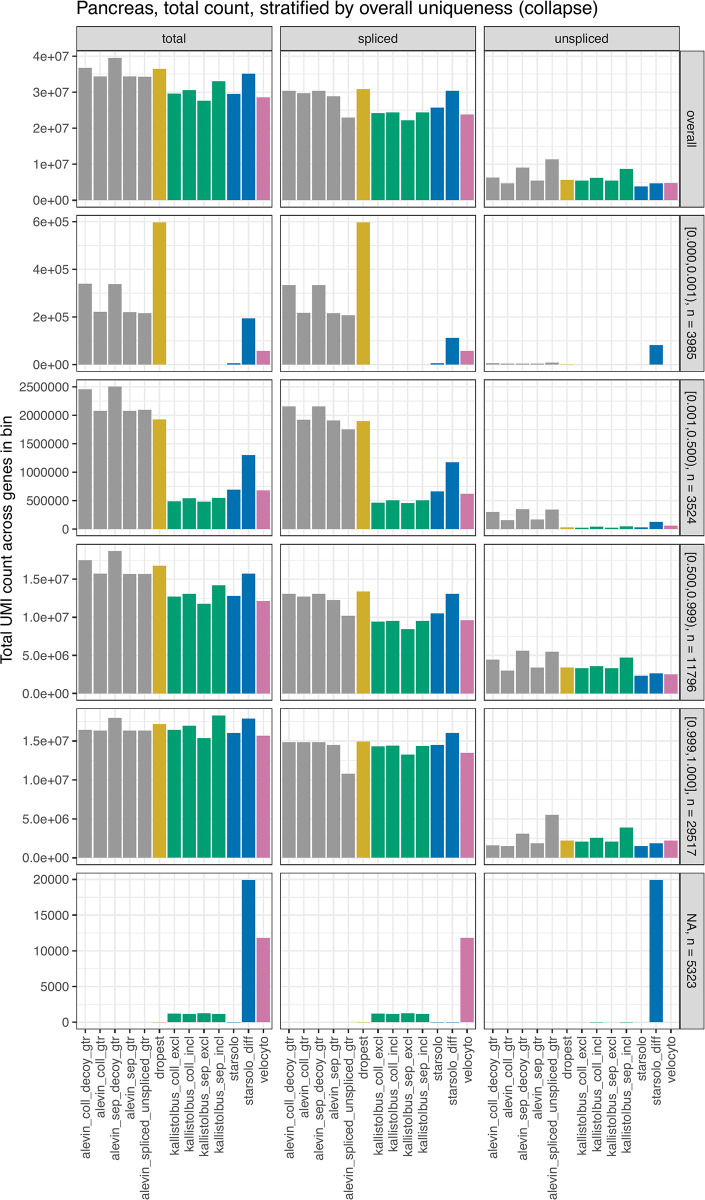
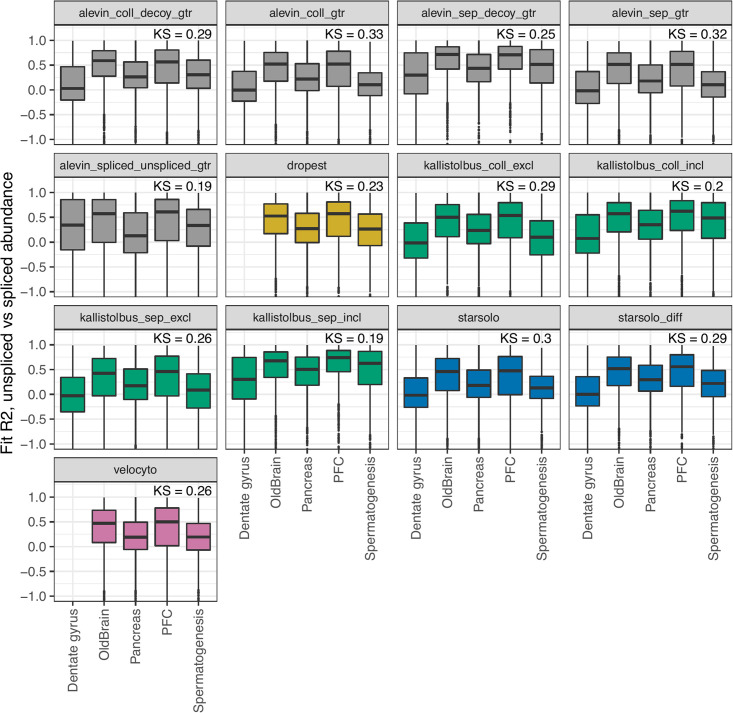
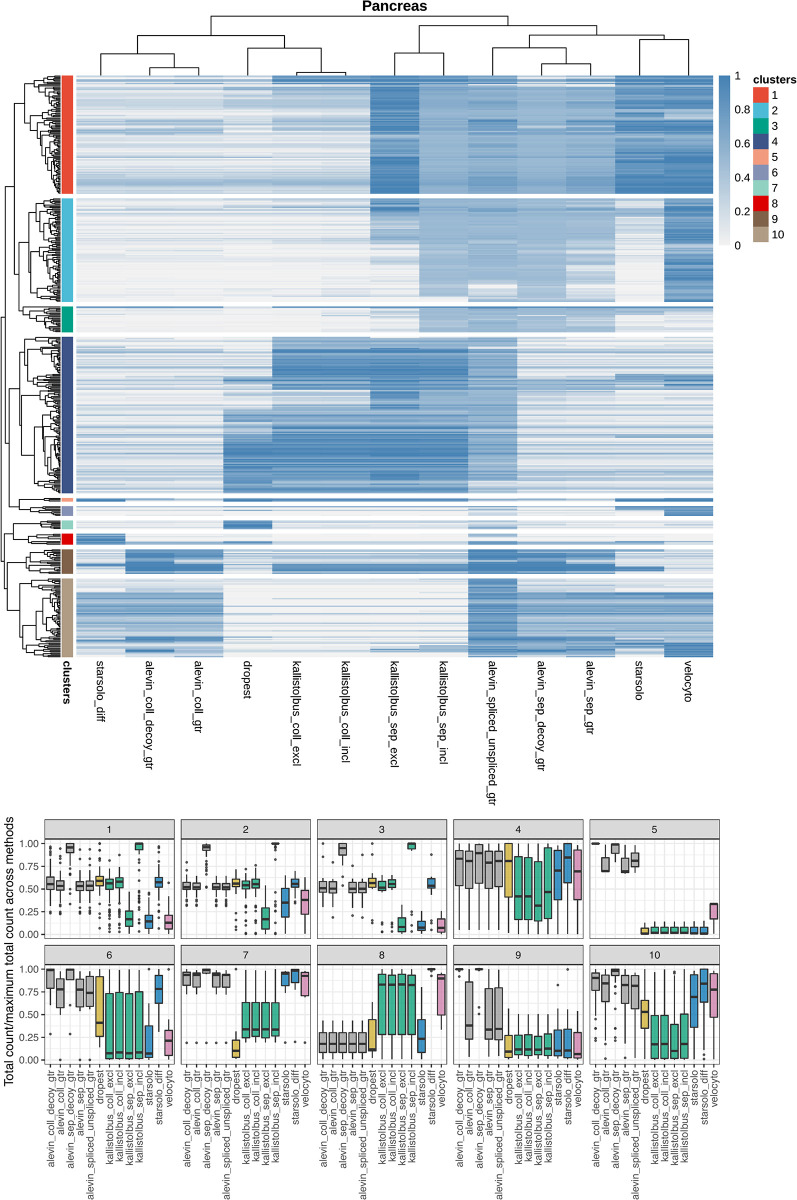
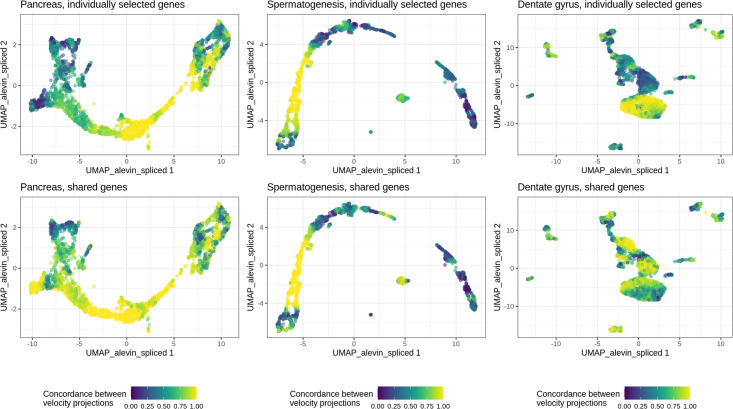
Experimental single-cell approaches are becoming widely used for many purposes, including investigation of the dynamic behaviour of developing biological systems. Consequently, a large number of computational methods for extracting dynamic information from such data have been developed. One example is RNA velocity analysis, in which spliced and unspliced RNA abundances are jointly modeled in order to infer a 'direction of change' and thereby a future state for each cell in the gene expression space. Naturally, the accuracy and interpretability of the inferred RNA velocities depend crucially on the correctness of the estimated abundances. Here, we systematically compare five widely used quantification tools, in total yielding thirteen different quantification approaches, in terms of their estimates of spliced and unspliced RNA abundances in five experimental droplet scRNA-seq data sets. We show that there are substantial differences between the quantifications obtained from different tools, and identify typical genes for which such discrepancies are observed. We further show that these abundance differences propagate to the downstream analysis, and can have a large effect on estimated velocities as well as the biological interpretation. Our results highlight that abundance quantification is a crucial aspect of the RNA velocity analysis workflow, and that both the definition of the genomic features of interest and the quantification algorithm itself require careful consideration.
实验单细胞方法正被广泛应用于多种目的,包括研究生物系统的动态行为。因此,已经开发出了大量用于从这些数据中提取动态信息的计算方法。一个例子是 RNA 速度分析,它联合建模了剪接和非剪接 RNA 的丰度,以推断每个细胞在基因表达空间中的“变化方向”,从而推断出其未来的状态。自然地,推断出的 RNA 速度的准确性和可解释性在很大程度上取决于估计丰度的正确性。在这里,我们系统性地比较了五个广泛使用的定量工具,总共产生了十三种不同的定量方法,根据它们在五个实验液滴 scRNA-seq 数据集的剪接和非剪接 RNA 丰度的估计。我们表明,不同工具获得的定量之间存在很大差异,并确定了观察到这种差异的典型基因。我们进一步表明,这些丰度差异会传播到下游分析中,并对估计的速度以及生物解释产生很大影响。我们的结果强调了丰度定量是 RNA 速度分析工作流程的关键方面,并且感兴趣的基因组特征的定义和定量算法本身都需要仔细考虑。