Microbiology Department, Centre for Human Virology and Genomics, Nigerian Institute of Medical Research, Lagos, Nigeria.
Biochemistry Department, Nigerian Institute of Medical Research, Lagos, Nigeria.
PLoS One. 2021 Jan 11;16(1):e0243271. doi: 10.1371/journal.pone.0243271. eCollection 2021.
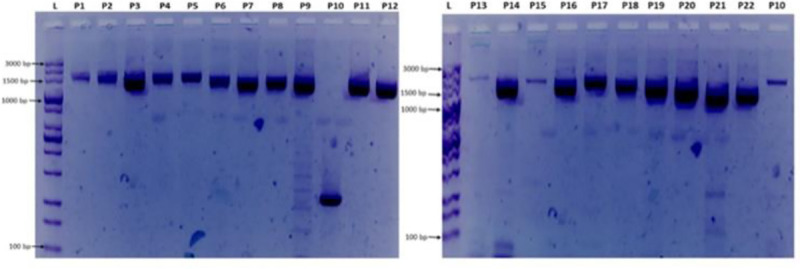
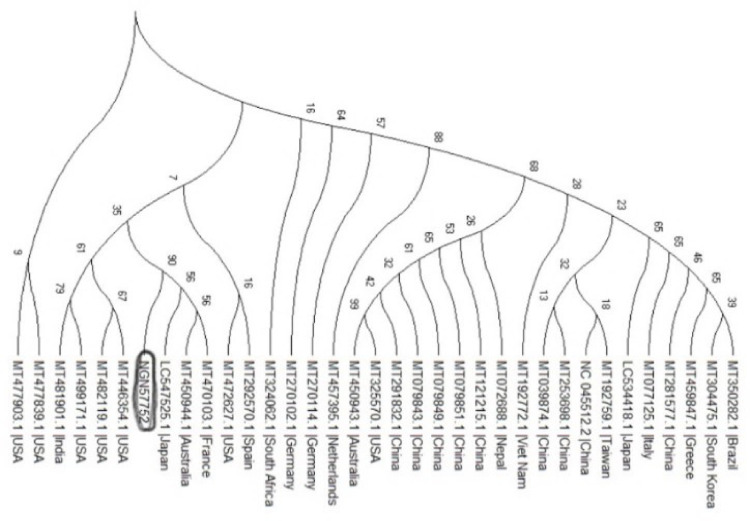
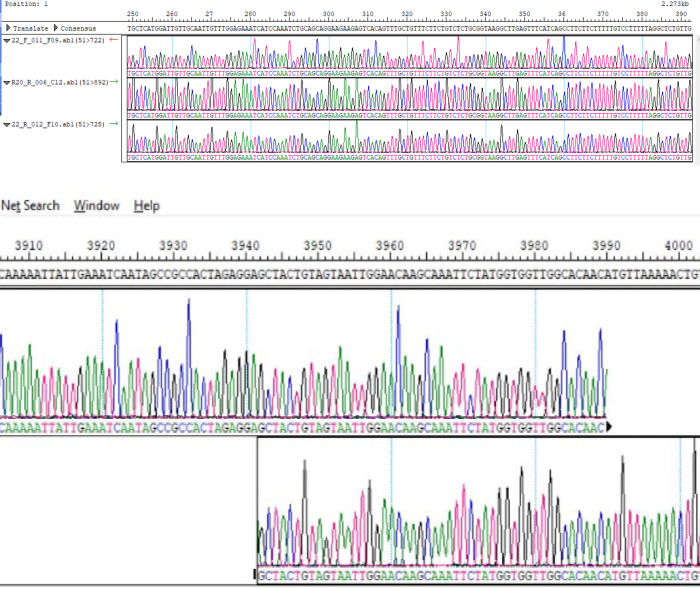
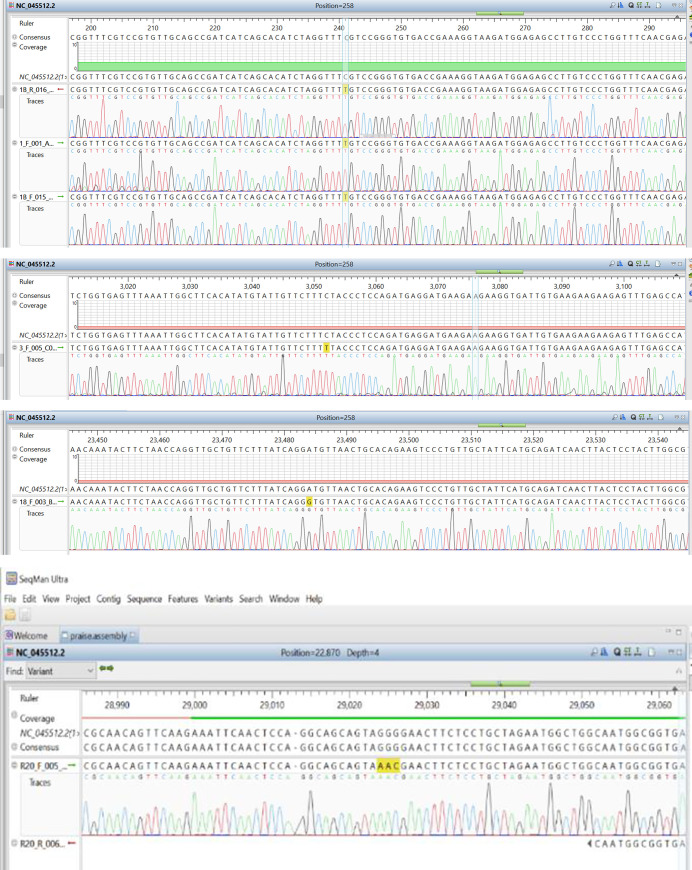
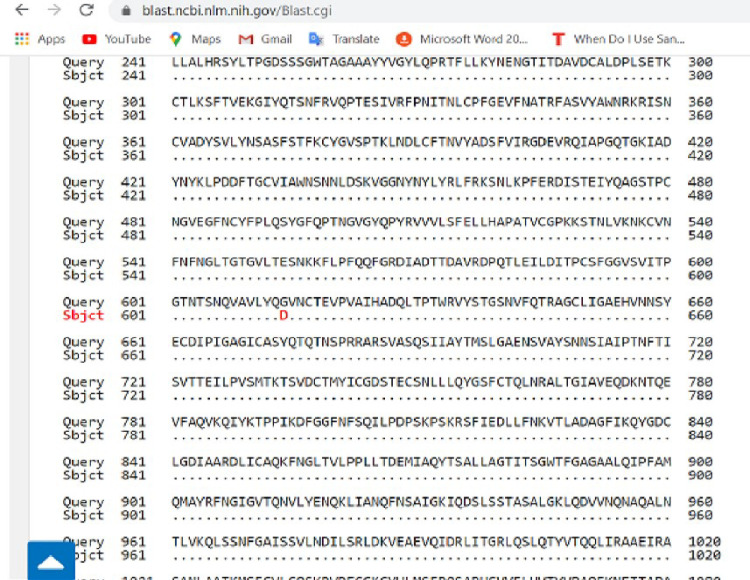
In an outbreak, effective detection of the aetiological agent(s) involved using molecular techniques is key to efficient diagnosis, early prevention and management of the spread. However, sequencing is necessary for mutation monitoring and tracking of clusters of transmission, development of diagnostics and for vaccines and drug development. Many sequencing methods are fast evolving to reduce test turn-around-time and to increase through-put compared to Sanger sequencing method; however, Sanger sequencing remains the gold standard for clinical research sequencing with its 99.99% accuracy This study sought to generate sequence data of SARS-CoV-2 using Sanger sequencing method and to characterize them for possible site(s) of mutations. About 30 pairs of primers were designed, synthesized, and optimized using endpoint PCR to generate amplicons for the full length of the virus. Cycle sequencing using BigDye Terminator v.3.1 and capillary gel electrophoresis on ABI 3130xl genetic analyser were performed according to the manufacturers' instructions. The sequence data generated were assembled and analysed for variations using DNASTAR Lasergene 17 SeqMan Ultra. Total length of 29,760bp of SARS-CoV-2 was assembled from the sample analysed and deposited in GenBank with accession number: MT576584. Blast result of the sequence assembly shows a 99.97% identity with the reference sequence. Variations were noticed at positions: nt201, nt2997, nt14368, nt16535, nt20334, and nt28841-28843, which caused amino acid alterations at the S (aa614) and N (aa203-204) regions. The mutations observed at S and N-gene in this study may be indicative of a gradual changes in the genetic coding of the virus hence, the need for active surveillance of the viral genome.
在疫情爆发期间,使用分子技术有效检测涉及的病原体对于高效诊断、早期预防和控制传播至关重要。然而,测序对于突变监测和传播群的追踪、诊断方法的开发以及疫苗和药物的开发都是必要的。与 Sanger 测序方法相比,许多测序方法正在快速发展,以缩短测试周转时间并提高通量;然而,Sanger 测序仍然是临床研究测序的金标准,其准确率为 99.99%。本研究旨在使用 Sanger 测序方法生成 SARS-CoV-2 的序列数据,并对其进行特征分析,以确定可能的突变位点。设计、合成了约 30 对引物,并使用终点 PCR 进行优化,以生成病毒全长的扩增子。根据制造商的说明,使用 BigDye Terminator v.3.1 进行循环测序,并在 ABI 3130xl 遗传分析仪上进行毛细管凝胶电泳。根据 DNASTAR Lasergene 17 SeqMan Ultra 对生成的序列数据进行组装和分析,以确定是否存在变异。从分析的样本中组装出全长 29760bp 的 SARS-CoV-2,并将其保存在 GenBank 中,登录号为:MT576584。序列组装的 Blast 结果显示与参考序列的同一性为 99.97%。在位置 nt201、nt2997、nt14368、nt16535、nt20334 和 nt28841-28843 处发现了变异,导致 S(aa614)和 N(aa203-204)区域的氨基酸改变。本研究中在 S 和 N 基因观察到的突变可能表明病毒遗传编码的逐渐变化,因此需要对病毒基因组进行积极监测。