Department of Biochemistry, Vanderbilt University School of Medicine, Nashville, Tennessee, USA.
Department of Biochemistry, Vanderbilt University School of Medicine, Nashville, Tennessee, USA.
J Biol Chem. 2021 Jan-Jun;296:100223. doi: 10.1074/jbc.RA120.016855. Epub 2020 Dec 25.
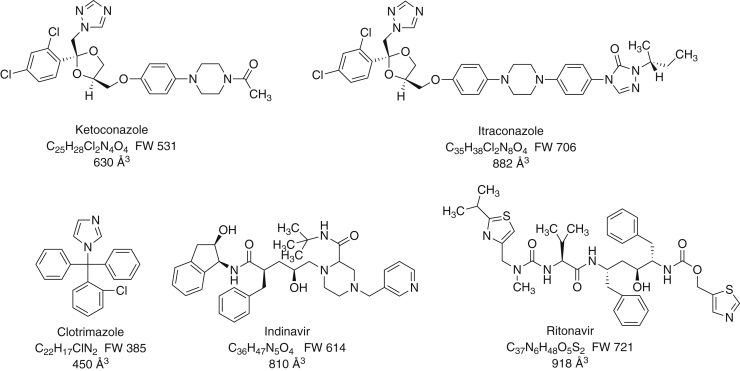
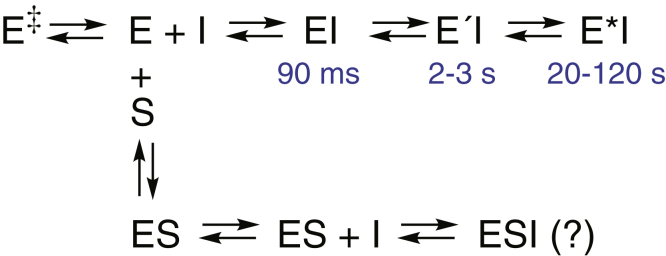
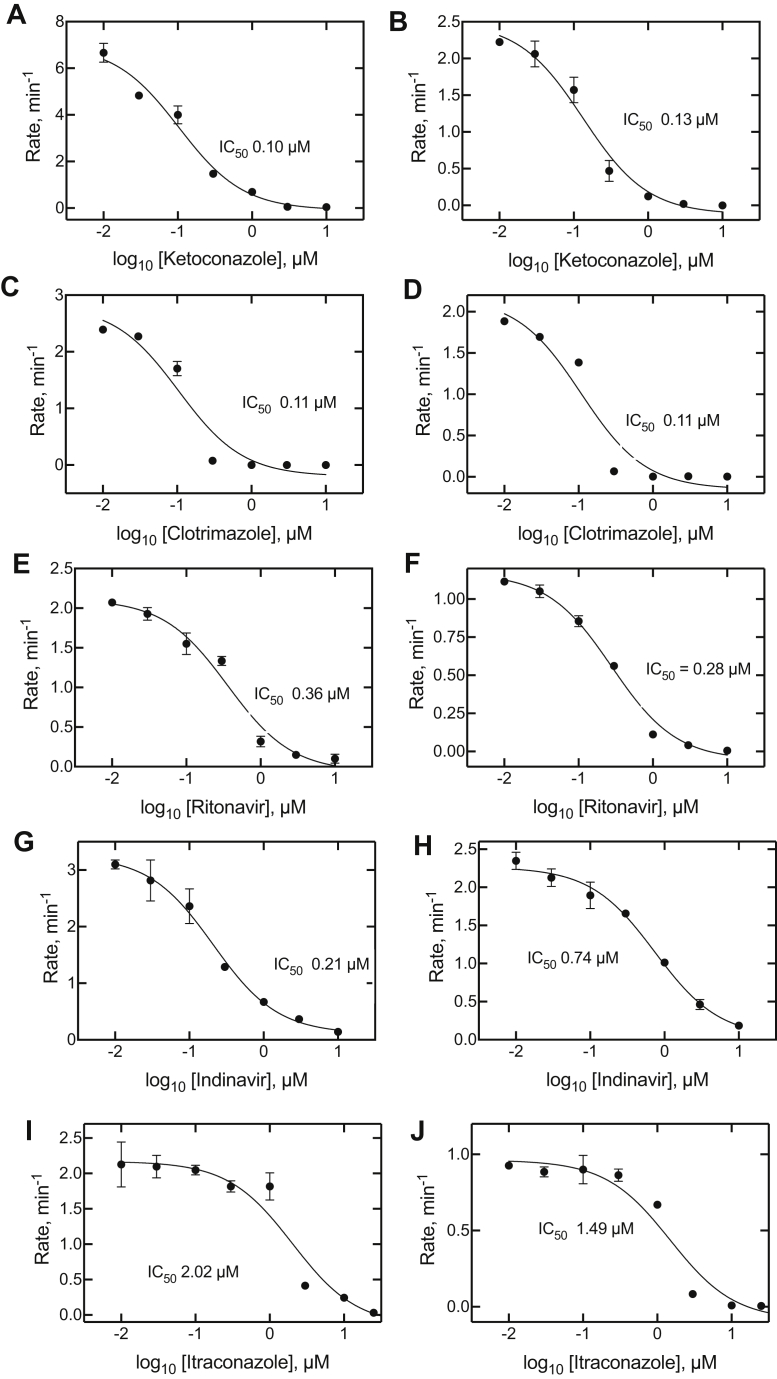
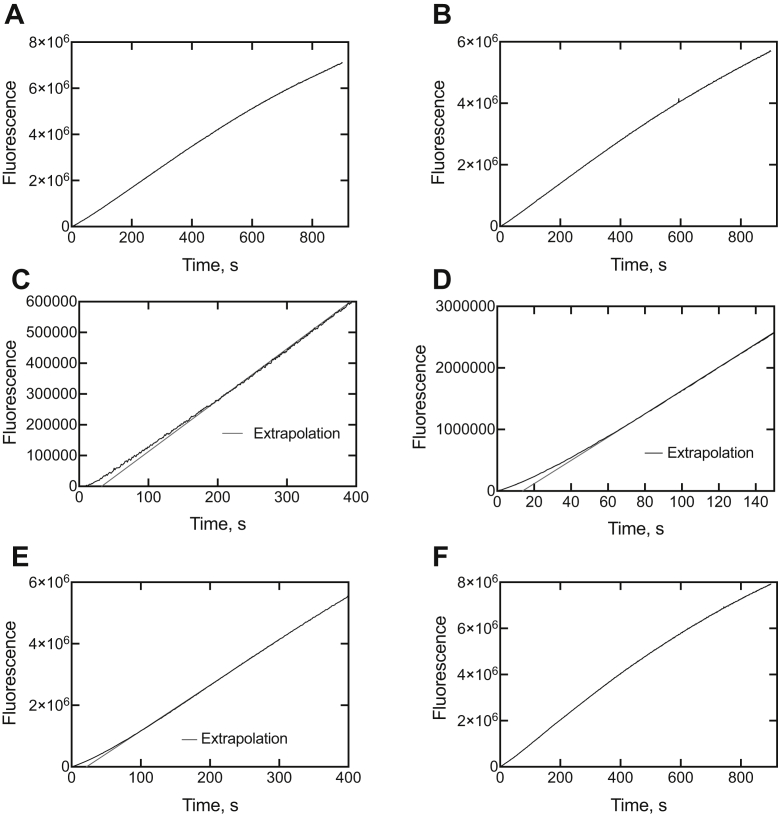
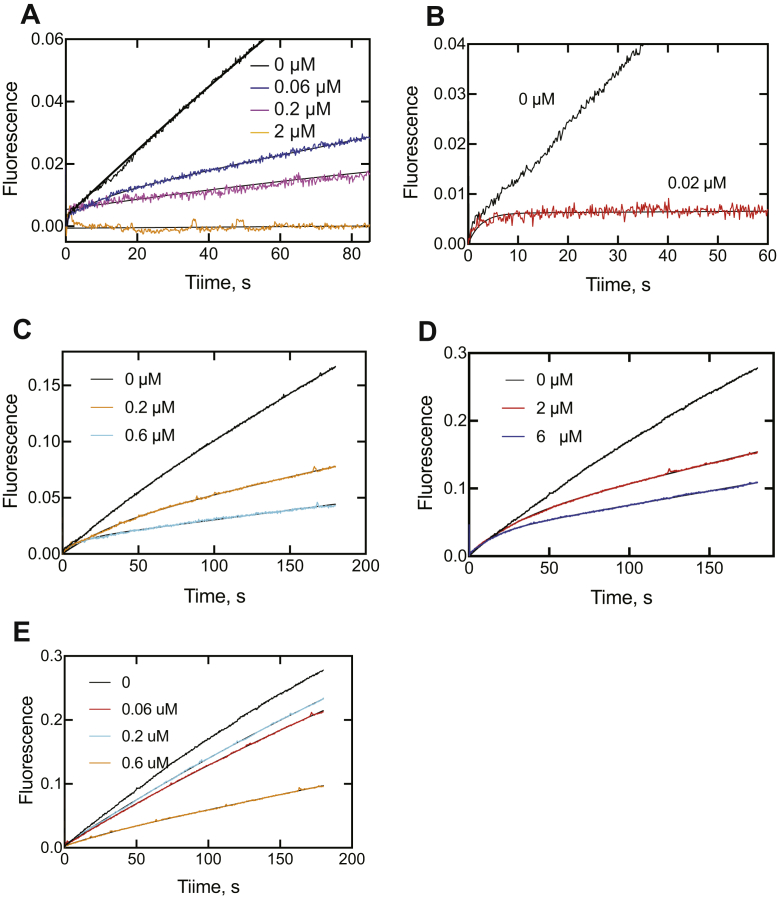
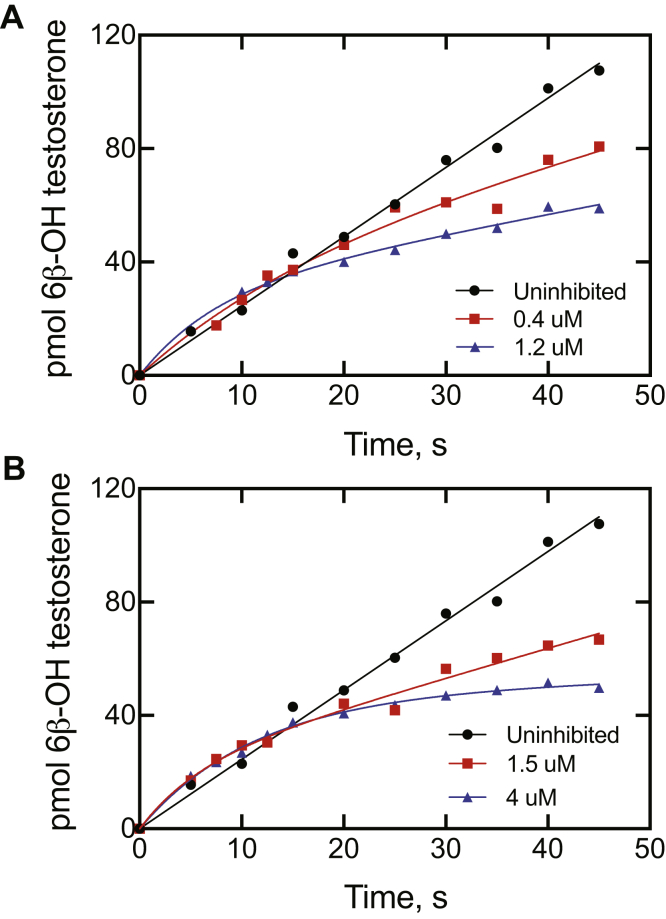
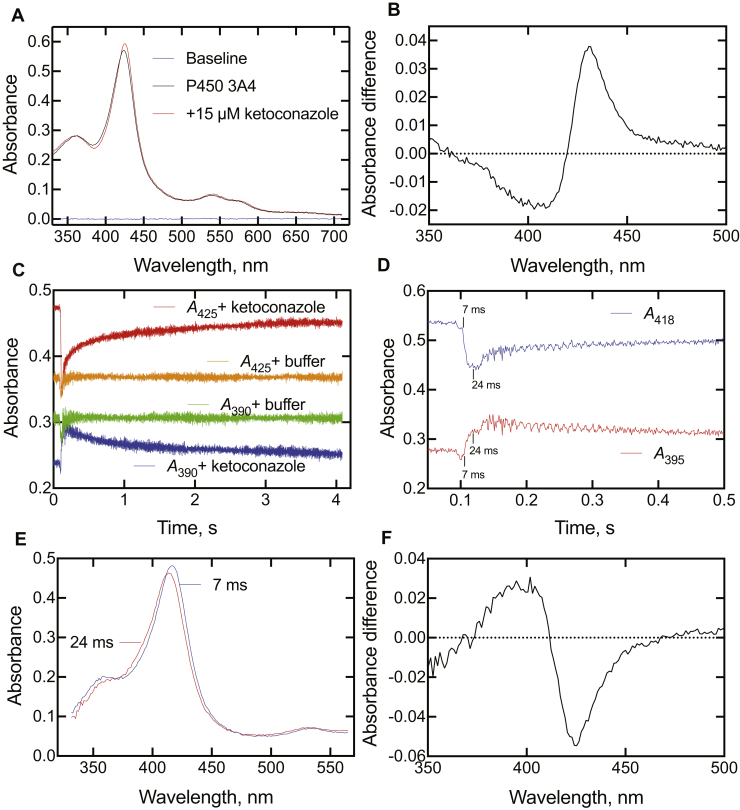
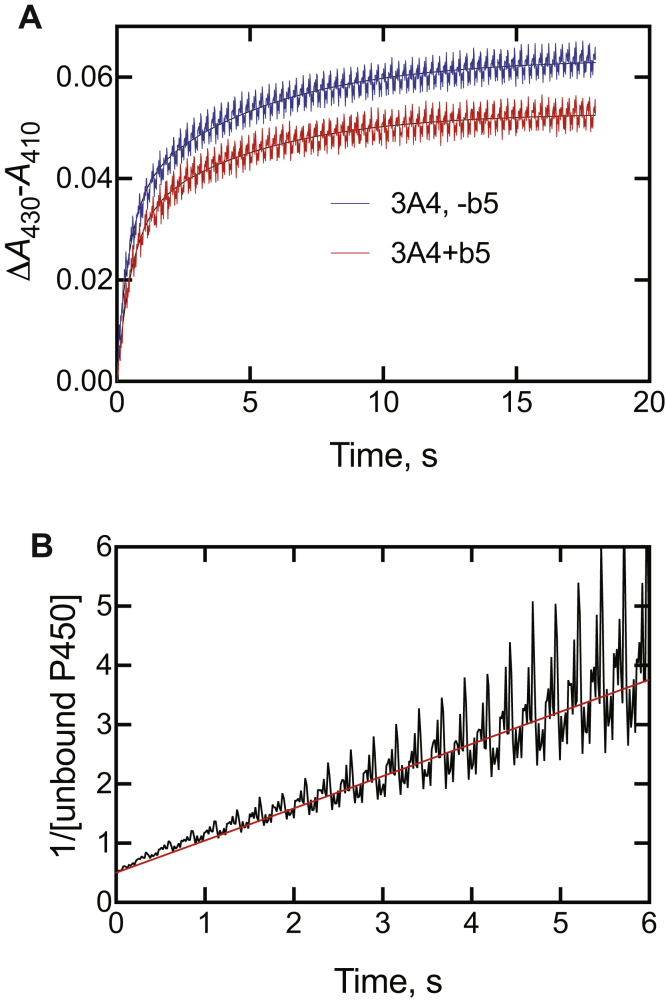
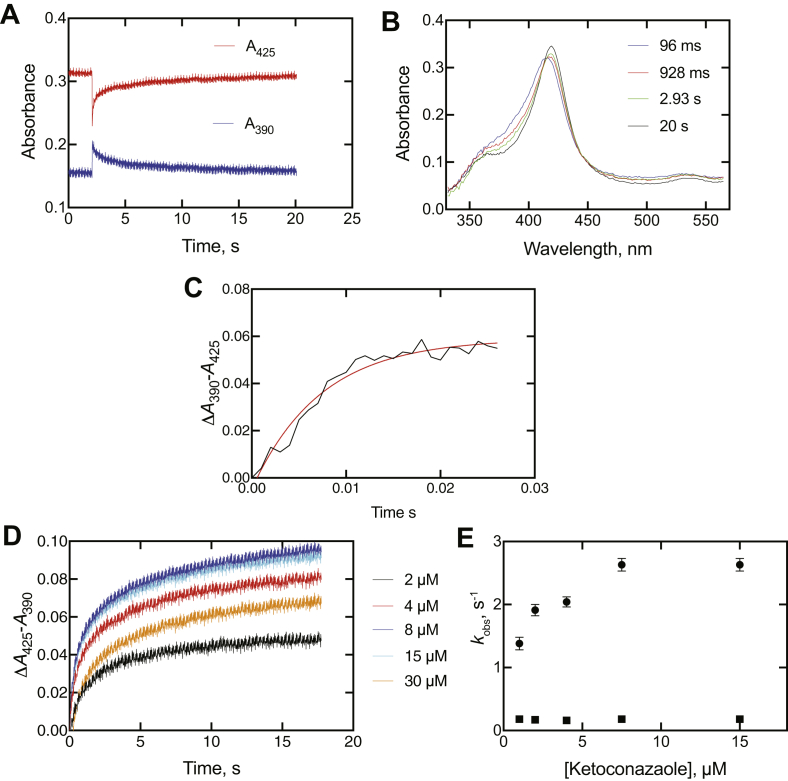
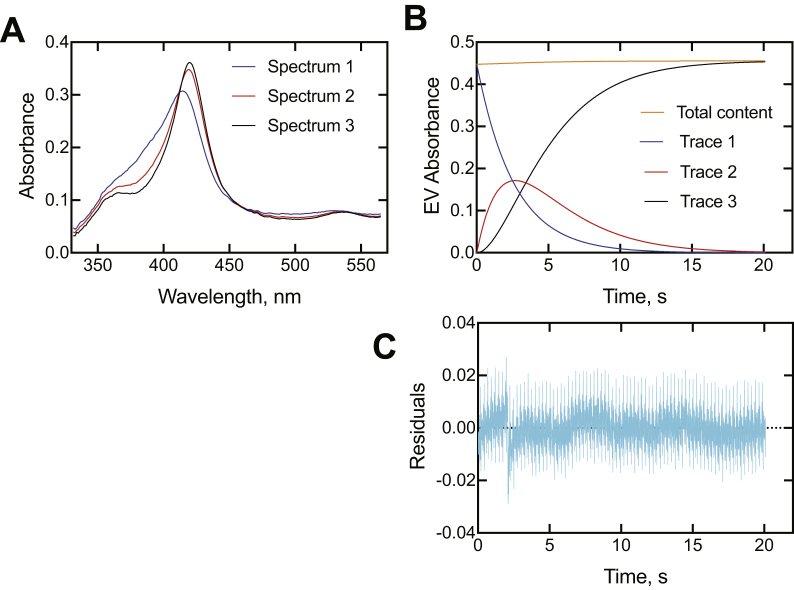
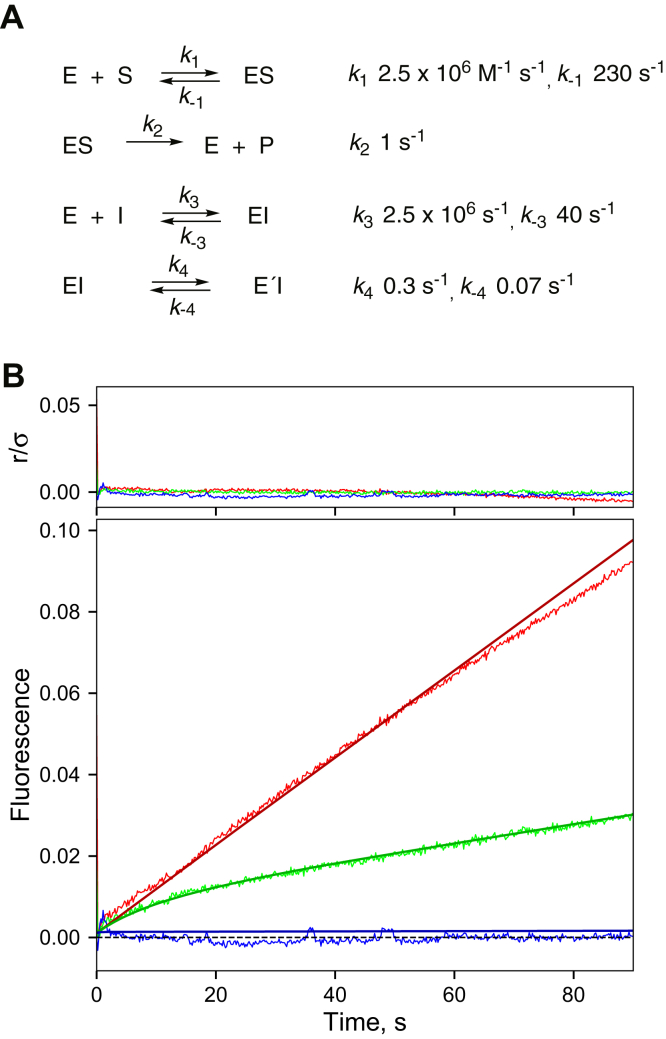
Cytochrome P450 (P450) 3A4 is the enzyme most involved in the metabolism of drugs and can also oxidize numerous steroids. This enzyme is also involved in one-half of pharmacokinetic drug-drug interactions, but details of the exact mechanisms of P450 3A4 inhibition are still unclear in many cases. Ketoconazole, clotrimazole, ritonavir, indinavir, and itraconazole are strong inhibitors; analysis of the kinetics of reversal of inhibition with the model substrate 7-benzoyl quinoline showed lag phases in several cases, consistent with multiple structures of P450 3A4 inhibitor complexes. Lags in the onset of inhibition were observed when inhibitors were added to P450 3A4 in 7-benzoyl quinoline O-debenzylation reactions, and similar patterns were observed for inhibition of testosterone 6β-hydroxylation by ritonavir and indinavir. Upon mixing with inhibitors, P450 3A4 showed rapid binding as judged by a spectral shift with at least partial high-spin iron character, followed by a slower conversion to a low-spin iron-nitrogen complex. The changes were best described by two intermediate complexes, one being a partial high-spin form and the second another intermediate, with half-lives of seconds. The kinetics could be modeled in a system involving initial loose binding of inhibitor, followed by a slow step leading to a tighter complex on a multisecond time scale. Although some more complex possibilities cannot be dismissed, these results describe a system in which conformationally distinct forms of P450 3A4 bind inhibitors rapidly and two distinct P450-inhibitor complexes exist en route to the final enzyme-inhibitor complex with full inhibitory activity.
细胞色素 P450(P450)3A4 是参与药物代谢的主要酶,也可以氧化许多甾体。这种酶还参与了一半的药代动力学药物相互作用,但在许多情况下,P450 3A4 抑制的确切机制的细节仍不清楚。酮康唑、克霉唑、利托那韦、茚地那韦和伊曲康唑是强抑制剂;用模型底物 7-苯甲酰喹啉分析抑制逆转的动力学表明,在几种情况下存在滞后阶段,与 P450 3A4 抑制剂复合物的多种结构一致。当抑制剂被添加到 7-苯甲酰喹啉 O-去苄化反应中的 P450 3A4 中时,观察到抑制的起始滞后,并且对于利托那韦和茚地那韦抑制睾酮 6β-羟化也观察到类似的模式。在与抑制剂混合时,P450 3A4 表现出快速结合,这可以通过光谱移位来判断,至少部分具有高自旋铁特性,随后缓慢转化为低自旋铁-氮络合物。这些变化最好用两个中间络合物来描述,一个是部分高自旋形式,另一个是另一个中间络合物,半衰期为秒。动力学可以用一个系统来建模,该系统涉及抑制剂的初始松散结合,然后是一个缓慢的步骤,导致在多毫秒时间尺度上形成更紧密的络合物。尽管不能排除一些更复杂的可能性,但这些结果描述了一个系统,其中 P450 3A4 的构象不同的形式快速结合抑制剂,并且在最终的酶-抑制剂复合物具有完全抑制活性之前存在两种不同的 P450-抑制剂络合物。