Institute for Translational Research in Biomedicine, Kaohsiung Chang Gung Memorial Hospital, Kaohsiung, Taiwan.
Institute of Biomedical Sciences, Academia Sinica, Taipei, Taiwan.
Theranostics. 2021 Jan 1;11(6):2594-2611. doi: 10.7150/thno.51648. eCollection 2021.
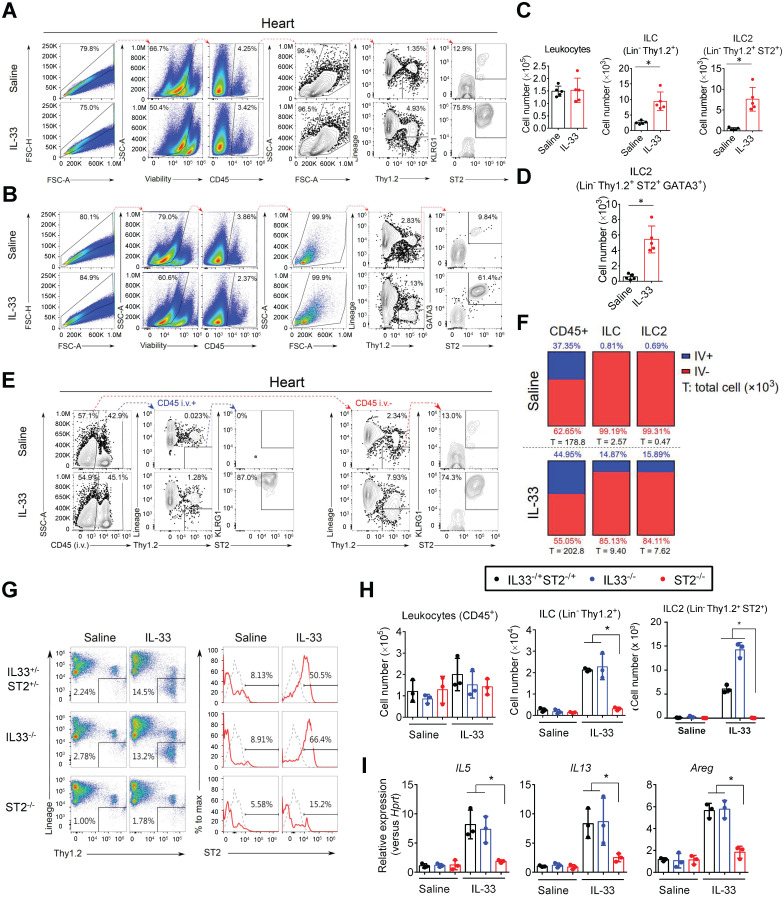
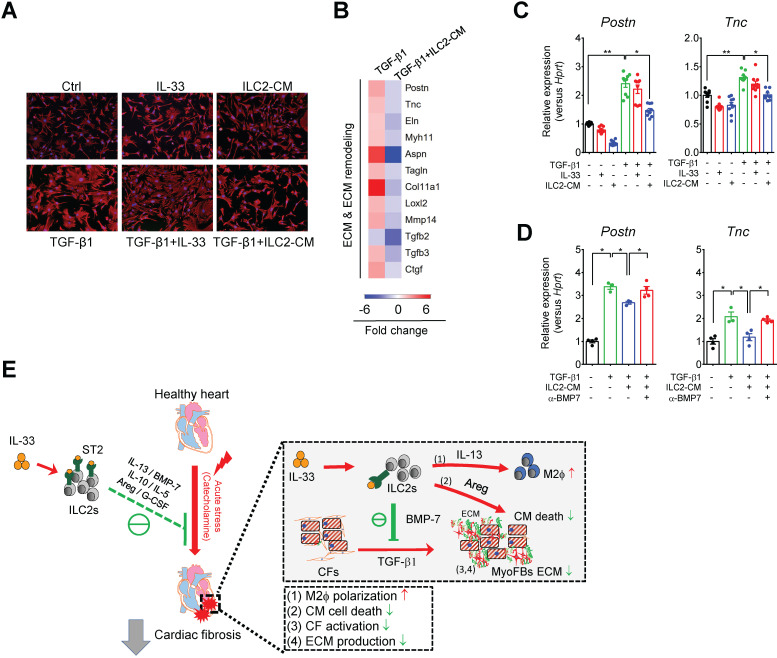
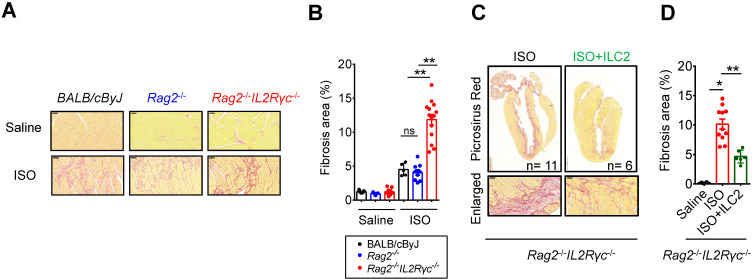
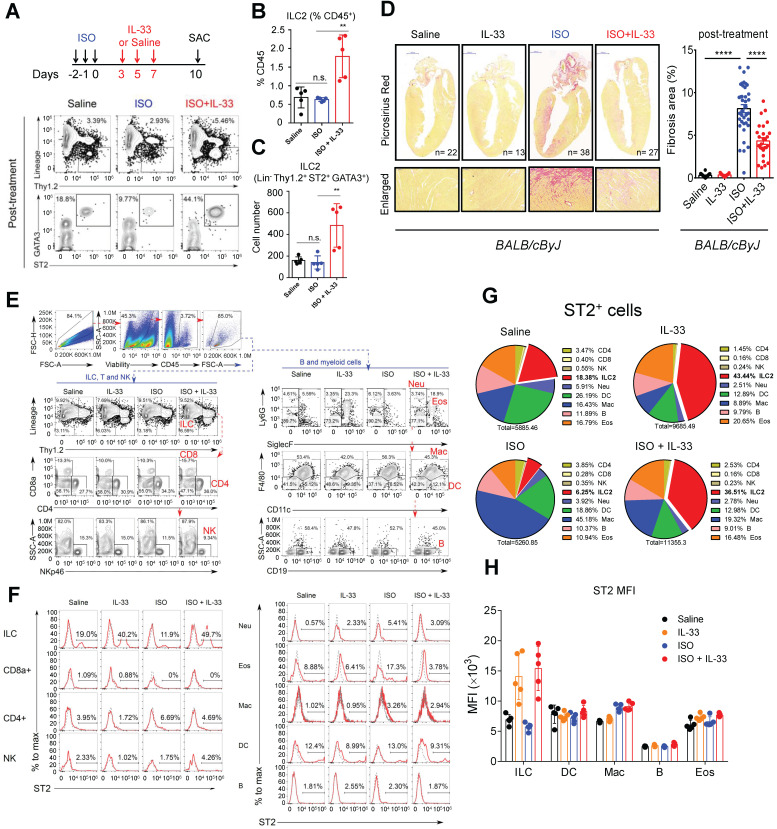
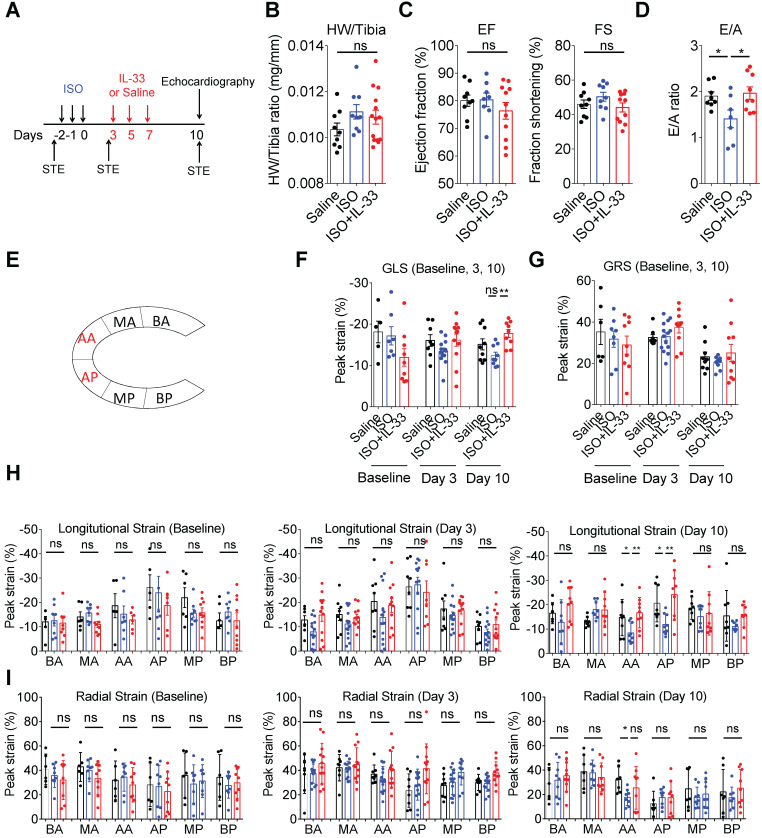
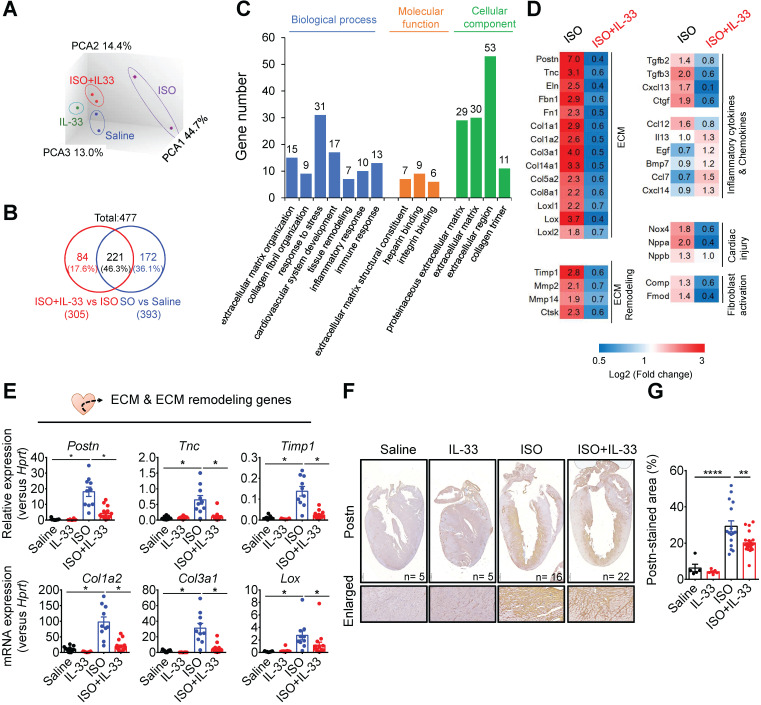
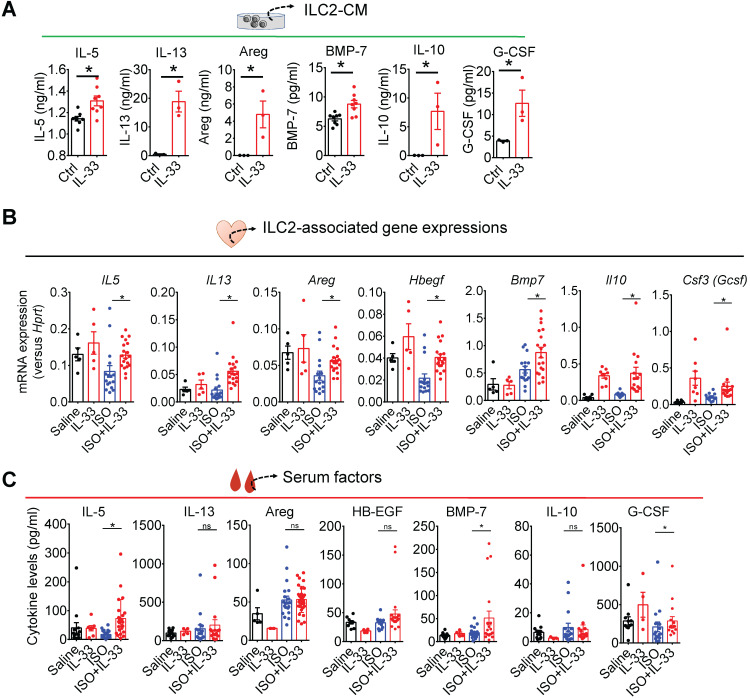
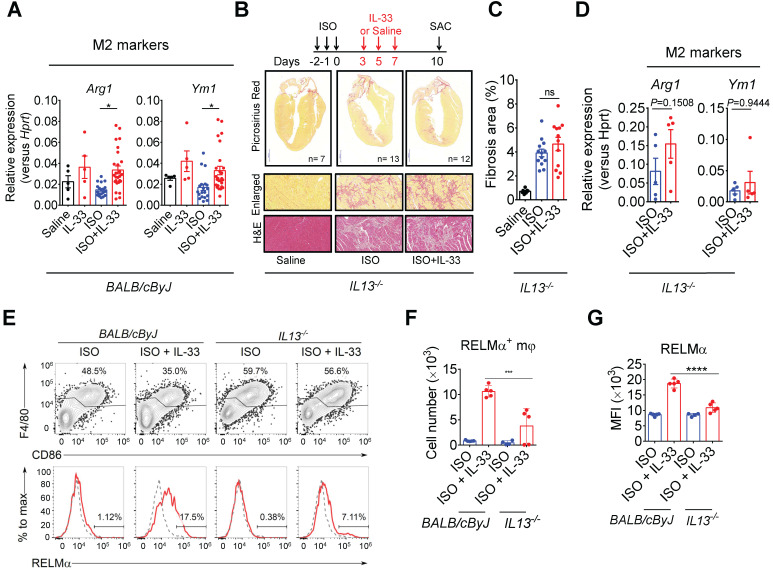
The major cause of heart failure is myocardium death consequent to detrimental cardiac remodeling and fibrosis following myocardial infarction. The cardiac protective cytokine interleukin (IL)-33, which signals by ST2 receptor binding, is associated with group 2 innate lymphoid cell (ILC2) activation and regulates tissue homeostasis and repair following tissue injury in various tissues. However, the distribution and role of IL-33-responsive ILC2s in cardiac fibrosis remain unclear. In this study, we elucidated the roles of IL-33-responsive cardiac-resident ILC2s and IL-33-mediated immunomodulatory functions in cardiac fibrosis. We examined the distribution of cardiac ILC2s by using flow cytometry. The roles of IL-33-mediated ILC2 expansion in cardiac fibrosis was evaluated in the mouse model of catecholamine-induced cardiac fibrosis. ILC-deficient mice were implemented to determine the contribution of endogenous ILC in the progression of cardiac fibrosis. Histopathological assessments, speckle tracking echocardiography, and transcriptome profile analysis were performed to determine the effects of IL-33-mediated cardiac protective functions. We identified the resident cardiac ILC2s, which share similar cell surface marker and transcriptional factor expression characteristics as peripheral blood and lung tissue ILC2s. IL-33 treatment induced ILC2 expansion via ST2. , ILC-deficient mice developed exacerbated cardiac fibrosis following catecholamine-induced stress cardiac injury. IL-33 treatment expanded cardiac ILC2s and revealed protective effects against cardiac tissue damage with reduced cardiomyocyte death, immune cell infiltration, tissue fibrosis, and improved myocardial function. Transcriptome analysis revealed that IL-33 attenuated extracellular matrix synthesis- and fibroblast activation-associated gene expressions. -knockout or epidermal growth factor receptor (EGFR) inhibition abolished IL-33-mediated cardiac protective function, confirming IL-13 and EGFR signaling as crucial for IL-33-mediated cardioprotective responses. Moreover, ILC2-produced BMP-7 served as a novel anti-fibrotic factor to inhibit TGF-β1-induced cardiac fibroblast activation. Our findings indicate the presence of IL-33-responsive ILC2s in cardiac tissue and that IL-33-mediated ILC2 expansion affords optimal cardioprotective function via ILC2-derived factors. IL-33-mediated immunomodulation is thus a promising strategy to promote tissue repair and alleviate cardiac fibrosis following acute cardiac injury.
心力衰竭的主要原因是心肌梗塞后有害的心脏重构和纤维化导致的心肌死亡。心肌保护性细胞因子白细胞介素 (IL)-33 通过 ST2 受体结合信号传递,与 2 型固有淋巴细胞 (ILC2) 的激活有关,并在各种组织损伤后调节组织稳态和修复。然而,IL-33 反应性 ILC2 在心脏纤维化中的分布和作用仍不清楚。在这项研究中,我们阐明了 IL-33 反应性心脏驻留 ILC2 和 IL-33 介导的免疫调节功能在心脏纤维化中的作用。我们使用流式细胞术检查了心脏 ILC2 的分布。在儿茶酚胺诱导的心脏纤维化的小鼠模型中评估了 IL-33 介导的 ILC2 扩张在心脏纤维化中的作用。实施了 ILC 缺陷小鼠以确定内源性 ILC 对心脏纤维化进展的贡献。进行了组织病理学评估、斑点跟踪超声心动图和转录组谱分析,以确定 IL-33 介导的心脏保护功能的影响。我们鉴定了驻留的心脏 ILC2,它们具有与外周血和肺组织 ILC2 相似的细胞表面标记和转录因子表达特征。IL-33 治疗通过 ST2 诱导 ILC2 扩张。IL-33 缺陷小鼠在儿茶酚胺诱导的应激性心脏损伤后发生更严重的心脏纤维化。IL-33 治疗可扩张心脏 ILC2,并通过减少心肌细胞死亡、免疫细胞浸润、组织纤维化和改善心肌功能来显示对心脏组织损伤的保护作用。转录组分析显示,IL-33 可减弱细胞外基质合成和成纤维细胞激活相关基因的表达。IL-13 和表皮生长因子受体 (EGFR) 抑制消除了 IL-33 介导的心脏保护功能,证实了 IL-13 和 EGFR 信号传导对 IL-33 介导的心脏保护反应至关重要。此外,ILC2 产生的 BMP-7 作为一种新型抗纤维化因子,可抑制 TGF-β1 诱导的心脏成纤维细胞激活。我们的研究结果表明,心脏组织中存在 IL-33 反应性 ILC2,IL-33 介导的 ILC2 扩张通过 ILC2 衍生的因子提供最佳的心脏保护功能。因此,IL-33 介导的免疫调节是一种很有前途的策略,可以促进急性心脏损伤后的组织修复和减轻心脏纤维化。