Department of Central Laboratory, The Affiliated Huaian No. 1 People's Hospital, Nanjing Medical University, Huai'an, Jiangsu, 223300, China.
Department of Hematology, The Affiliated Huaian No.1 People's Hospital,Nanjing Medical University, Huai'an, Jiangsu, 223300, China.
Theranostics. 2021 Jan 1;11(6):2691-2705. doi: 10.7150/thno.50571. eCollection 2021.
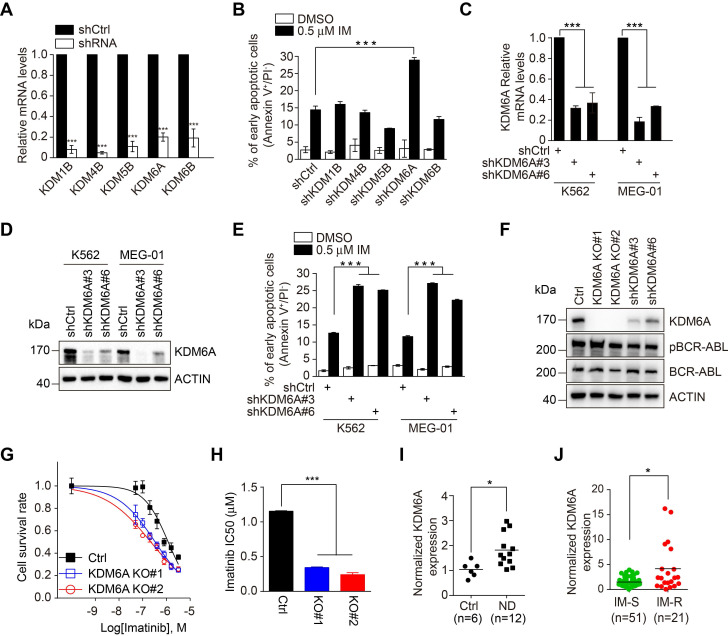
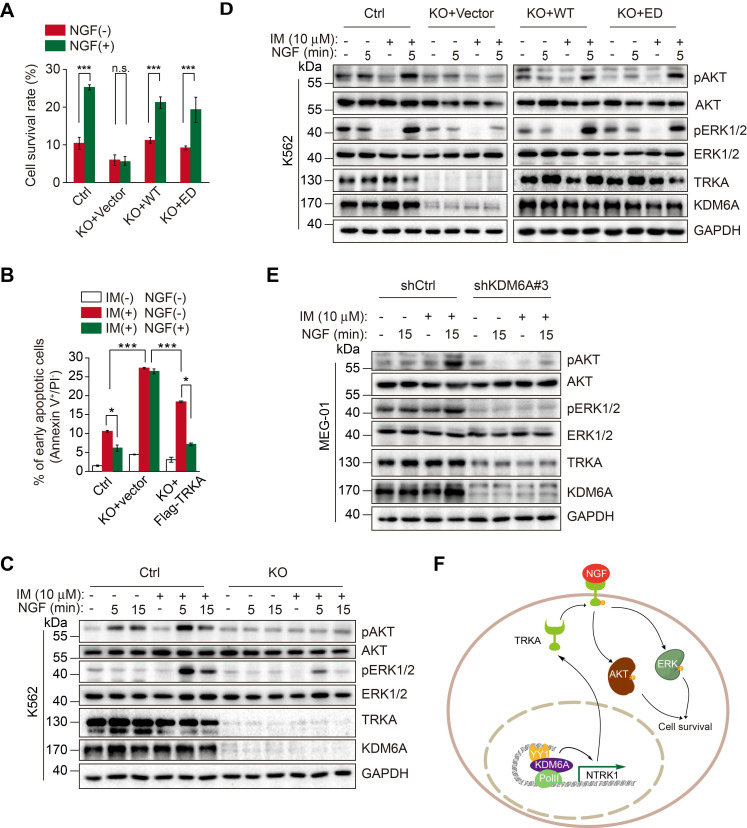
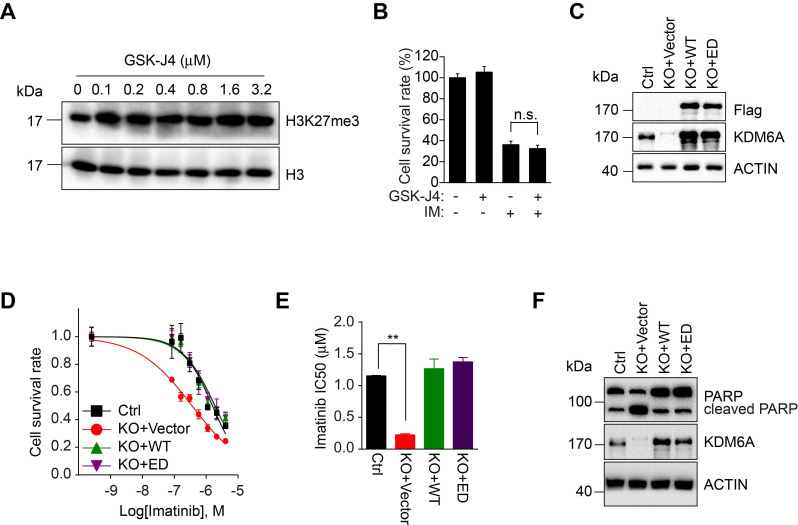
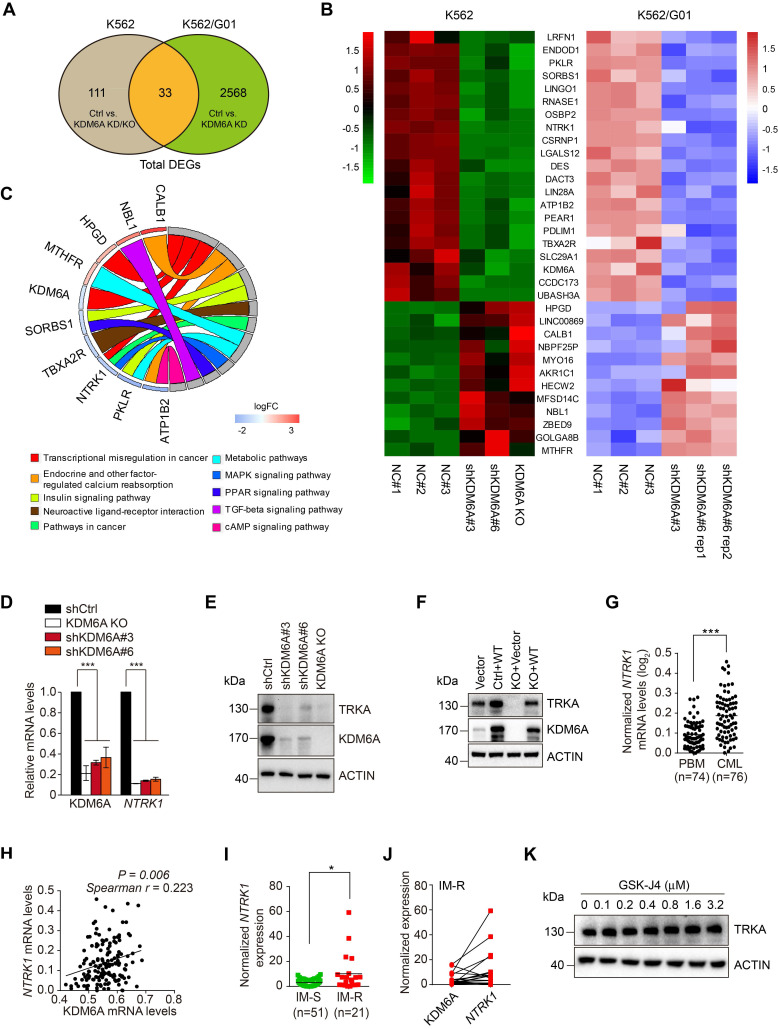
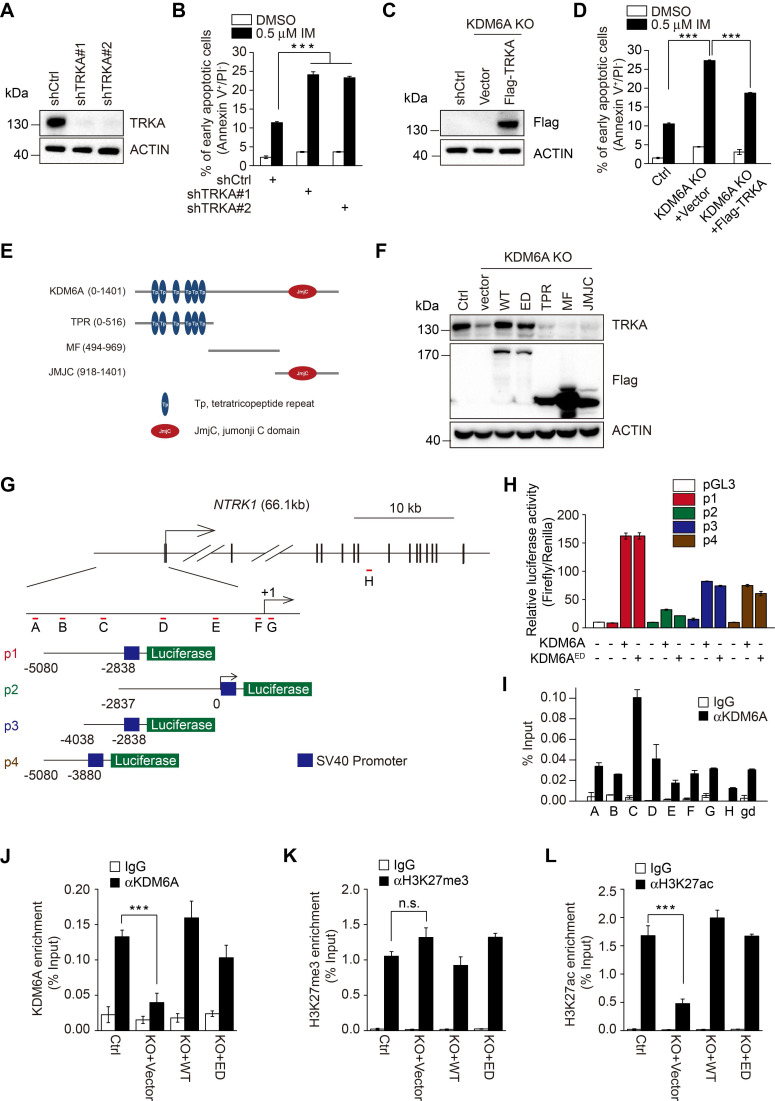
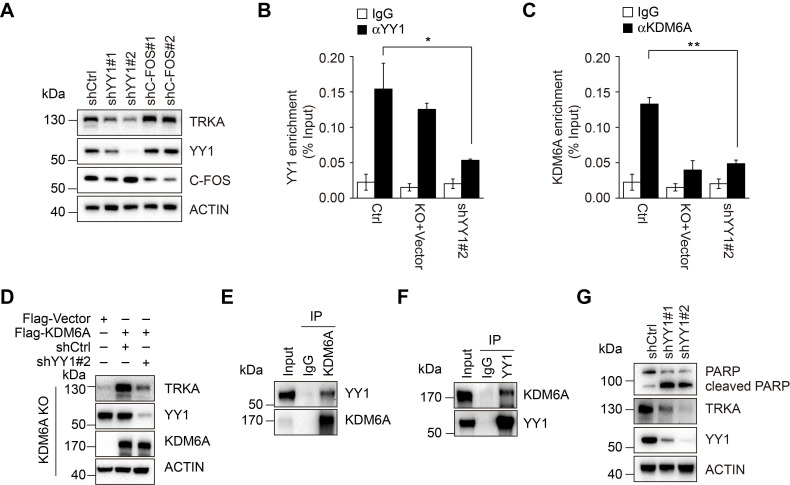
Despite landmark therapy of chronic myelogenous leukemia (CML) with tyrosine kinase inhibitors (TKIs), drug resistance remains problematic. Cancer pathogenesis involves epigenetic dysregulation and in particular, histone lysine demethylases (KDMs) have been implicated in TKI resistance. We sought to identify KDMs with altered expression in CML and define their contribution to imatinib resistance. Bioinformatics screening compared KDM expression in CML versus normal bone marrow with shRNA knockdown and flow cytometry used to measure effects on imatinib-induced apoptosis in K562 cells. Transcriptomic analyses were performed against KDM6A CRISPR knockout/shRNA knockdown K562 cells along with gene rescue experiments using wildtype and mutant demethylase-dead KDM6A constructs. Co-immunoprecipitation, luciferase reporter and ChIP were employed to elucidate mechanisms of KDM6A-dependent resistance. Amongst five KDMs upregulated in CML, only KDM6A depletion sensitized CML cells to imatinib-induced apoptosis. Re-introduction of demethylase-dead KDM6A as well as wild-type KDM6A restored imatinib resistance. RNA-seq identified NTRK1 gene downregulation after depletion of KDM6A. Moreover, NTRK1 expression positively correlated with KDM6A in a subset of clinical CML samples and KDM6A knockdown in fresh CML isolates decreased NTRK1 encoded protein (TRKA) expression. Mechanistically, KDM6A was recruited to the NTRK1 promoter by the transcription factor YY1 with subsequent TRKA upregulation activating down-stream survival pathways to invoke imatinib resistance. Contrary to its reported role as a tumor suppressor and independent of its demethylase function, KDM6A promotes imatinib-resistance in CML cells. The identification of the KDM6A/YY1/TRKA axis as a novel imatinib-resistance mechanism represents an unexplored avenue to overcome TKI resistance in CML.
尽管慢性髓系白血病 (CML) 的酪氨酸激酶抑制剂 (TKI) 治疗取得了里程碑式的进展,但耐药性仍然是一个问题。癌症的发病机制涉及表观遗传失调,特别是组蛋白赖氨酸去甲基酶 (KDMs) 已被牵连到 TKI 耐药性中。我们试图确定 CML 中表达改变的 KDMs,并定义它们对伊马替尼耐药性的贡献。 生物信息学筛选比较了 CML 与正常骨髓中的 KDM 表达,使用 shRNA 敲低,并使用流式细胞术测量对 K562 细胞中伊马替尼诱导凋亡的影响。对 KDM6A CRISPR 敲除/shRNA 敲低 K562 细胞进行转录组分析,并使用野生型和突变型去甲基酶失活 KDM6A 构建体进行基因挽救实验。共免疫沉淀、荧光素酶报告和 ChIP 用于阐明 KDM6A 依赖性耐药的机制。 在 CML 中上调的五 个 KDMs 中,只有 KDM6A 的耗竭使 CML 细胞对伊马替尼诱导的凋亡敏感。去甲基酶失活的 KDM6A 以及野生型 KDM6A 的重新引入恢复了伊马替尼的耐药性。RNA-seq 鉴定出 KDM6A 耗竭后 NTRK1 基因下调。此外,在一组临床 CML 样本中,NTRK1 的表达与 KDM6A 呈正相关,而新鲜 CML 分离物中的 KDM6A 敲低降低了 NTRK1 编码蛋白 (TRKA) 的表达。从机制上讲,转录因子 YY1 将 KDM6A 募集到 NTRK1 启动子,随后 TRKA 上调激活下游存活途径,引发伊马替尼耐药性。 与 KDM6A 作为肿瘤抑制因子的报道作用相反,并且与其去甲基酶功能无关,KDM6A 促进 CML 细胞对伊马替尼的耐药性。KDM6A/YY1/TRKA 轴作为一种新的伊马替尼耐药机制的鉴定代表了克服 CML 中 TKI 耐药性的一条未探索的途径。