Crowley Cheynna, Yang Yuchen, Qiu Yunjiang, Hu Benxia, Abnousi Armen, Lipiński Jakub, Plewczyński Dariusz, Wu Di, Won Hyejung, Ren Bing, Hu Ming, Li Yun
Department of Genetics, University of North Carolina Chapel Hill, Chapel Hill, NC, USA.
Department of Biostatistics, University of North Carolina Chapel Hill, Chapel Hill, NC, USA.
Comput Struct Biotechnol J. 2020 Dec 29;19:355-362. doi: 10.1016/j.csbj.2020.12.026. eCollection 2021.
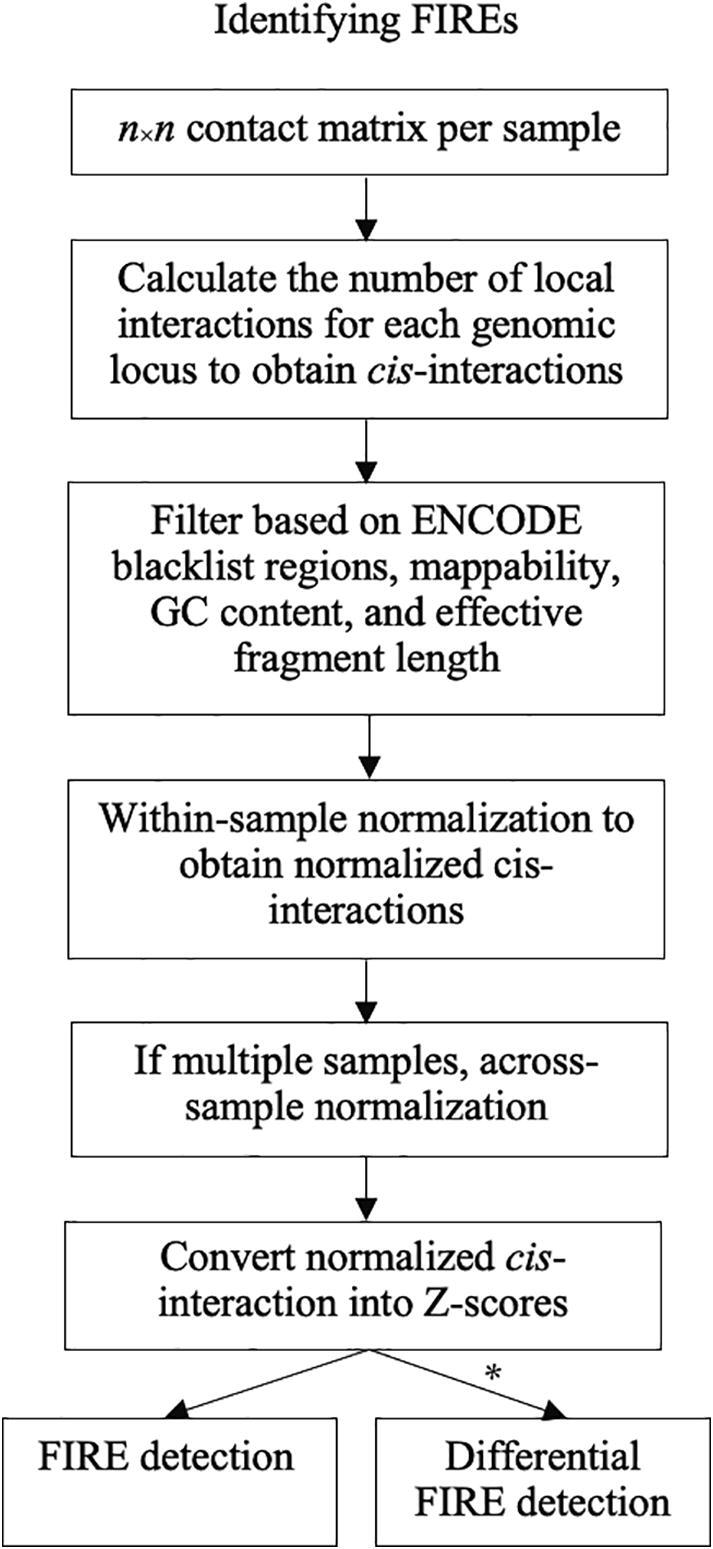
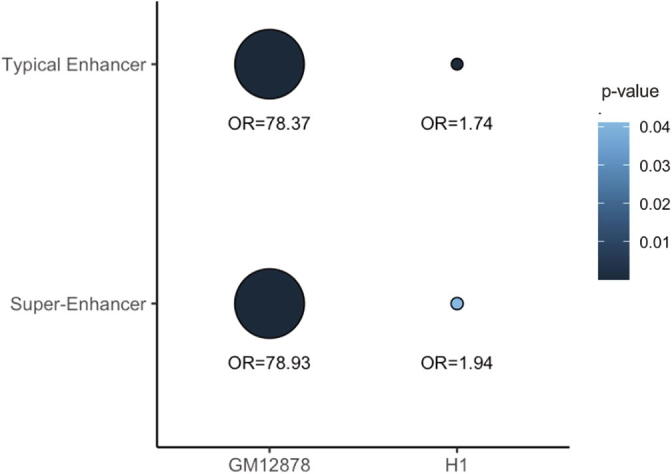
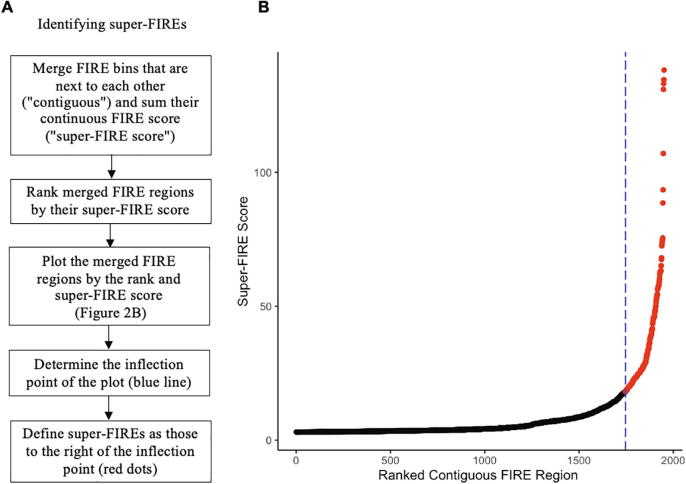
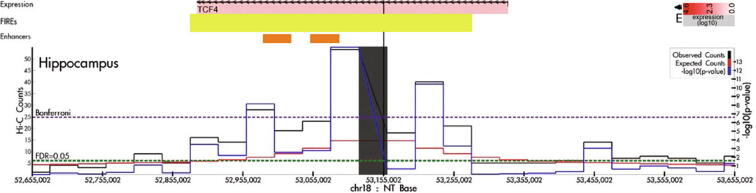
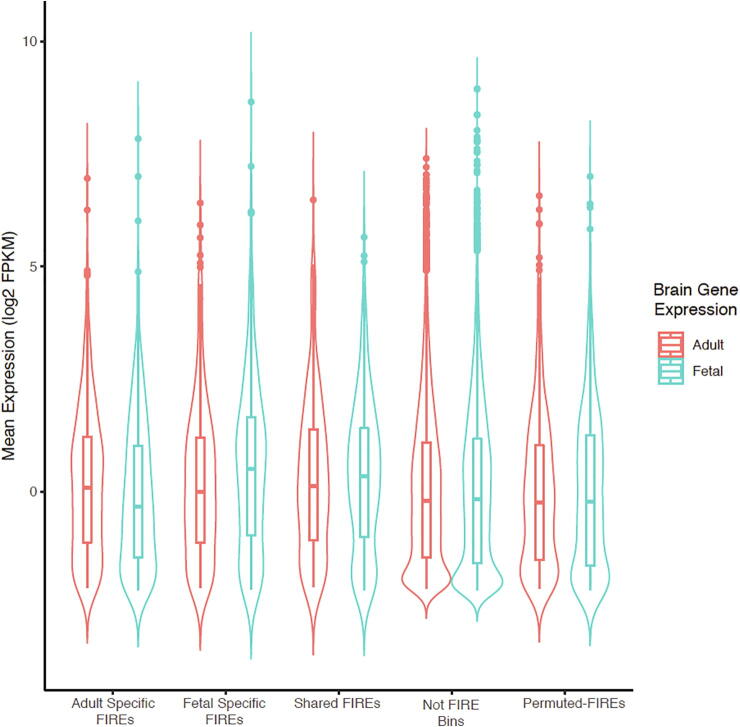
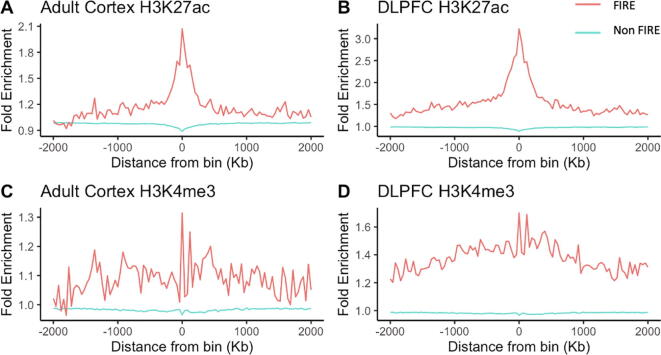
Hi-C experiments have been widely adopted to study chromatin spatial organization, which plays an essential role in genome function. We have recently identified frequently interacting regions (FIREs) and found that they are closely associated with cell-type-specific gene regulation. However, computational tools for detecting FIREs from Hi-C data are still lacking. In this work, we present FIREcaller, a stand-alone, user-friendly R package for detecting FIREs from Hi-C data. FIREcaller takes raw Hi-C contact matrices as input, performs within-sample and cross-sample normalization, and outputs continuous FIRE scores, dichotomous FIREs, and super-FIREs. Applying FIREcaller to Hi-C data from various human tissues, we demonstrate that FIREs and super-FIREs identified, in a tissue-specific manner, are closely related to gene regulation, are enriched for enhancer-promoter (E-P) interactions, tend to overlap with regions exhibiting epigenomic signatures of -regulatory roles, and aid the interpretation or GWAS variants. The FIREcaller package is implemented in R and freely available at https://yunliweb.its.unc.edu/FIREcaller.
Hi-C实验已被广泛用于研究染色质空间组织,其在基因组功能中起着至关重要的作用。我们最近鉴定出了频繁相互作用区域(FIREs),并发现它们与细胞类型特异性基因调控密切相关。然而,仍缺乏从Hi-C数据中检测FIREs的计算工具。在这项工作中,我们展示了FIREcaller,这是一个独立的、用户友好的R包,用于从Hi-C数据中检测FIREs。FIREcaller将原始Hi-C接触矩阵作为输入,进行样本内和跨样本归一化,并输出连续的FIRE分数、二分法FIREs和超级FIREs。将FIREcaller应用于来自各种人类组织的Hi-C数据,我们证明以组织特异性方式鉴定出的FIREs和超级FIREs与基因调控密切相关,富含增强子-启动子(E-P)相互作用,倾向于与表现出调控作用表观基因组特征的区域重叠,并有助于解释全基因组关联研究(GWAS)变体。FIREcaller包用R语言实现,可在https://yunliweb.its.unc.edu/FIREcaller上免费获取。