Gene Center, Ludwig-Maximilians-Universität München, Munich, 81377, Germany.
Google, Seattle, WA, 98103, United States.
Bioinformatics. 2021 Aug 4;37(14):2004–2011. doi: 10.1093/bioinformatics/btab050. Epub 2021 Jan 30.
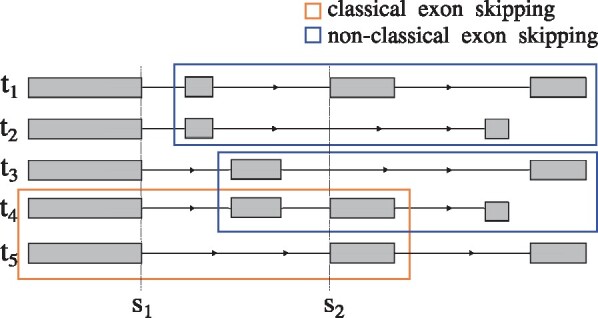
Alternative splicing removes intronic sequences from pre-mRNAs in alternative ways to produce different forms (isoforms) of mature mRNA. The composition of expressed transcripts gives specific functionalities to cells in a particular condition or developmental stage. In addition, a large fraction of human disease mutations affect splicing and lead to aberrant mRNA and protein products. Current methods that interrogate the transcriptome based on RNA-seq either suffer from short read length when trying to infer full-length transcripts, or are restricted to predefined units of alternative splicing that they quantify from local read evidence.
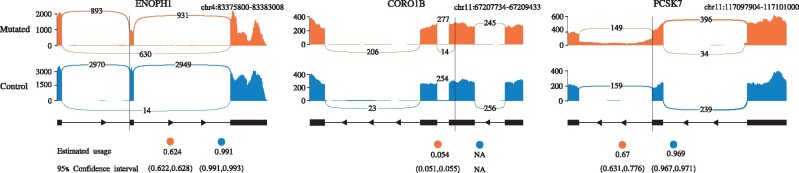
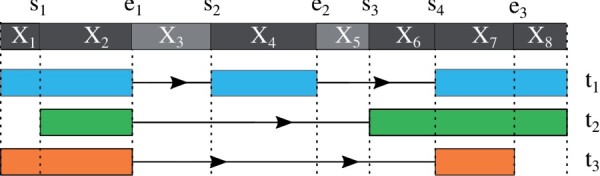
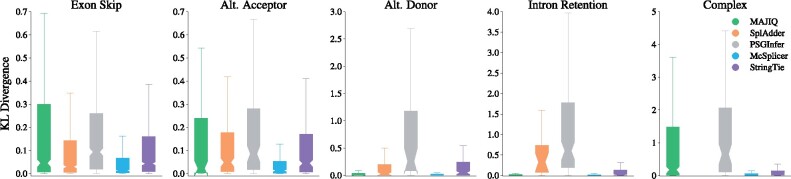
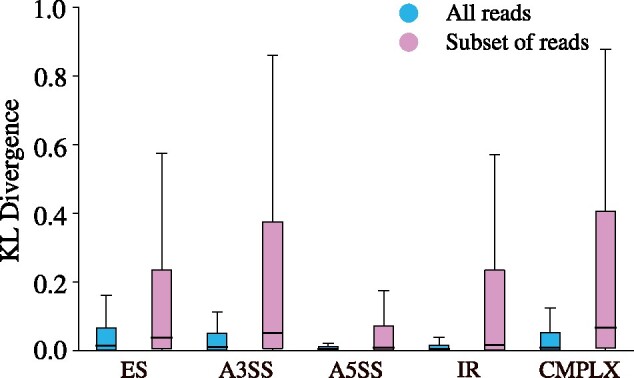
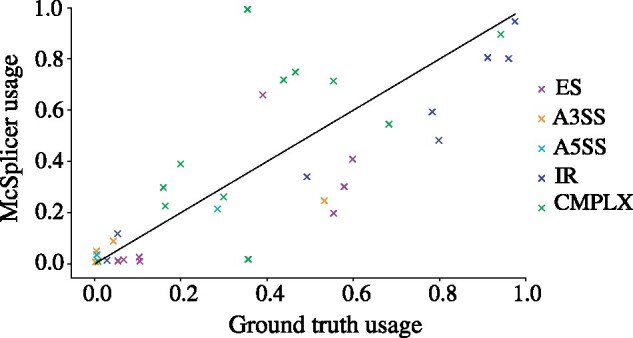
Instead of attempting to quantify individual outcomes of the splicing process such as local splicing events or full-length transcripts, we propose to quantify alternative splicing using a simplified probabilistic model of the underlying splicing process. Our model is based on the usage of individual splice sites and can generate arbitrarily complex types of splicing patterns. In our implementation, McSplicer, we estimate the parameters of our model using all read data at once and we demonstrate in our experiments that this yields more accurate estimates compared to competing methods. Our model is able to describe multiple effects of splicing mutations using few, easy to interpret parameters, as we illustrate in an experiment on RNA-seq data from autism spectrum disorder patients.
McSplicer source code is available at https://github.com/canzarlab/McSplicer and has been deposited in archived format at https://doi.org/10.5281/zenodo.4449881.
Supplementary data are available at Bioinformatics online.
可变剪接以不同的方式从前体 mRNA 中去除内含子序列,从而产生不同形式(异构体)的成熟 mRNA。表达转录本的组成赋予特定条件或发育阶段的细胞特定的功能。此外,很大一部分人类疾病突变影响剪接,导致异常的 mRNA 和蛋白质产物。目前基于 RNA-seq 检测转录组的方法在尝试推断全长转录本时,由于读长较短,或者仅限于它们从局部读码证据中定量的预定可变剪接单位。
我们不是试图量化剪接过程的单个结果,例如局部剪接事件或全长转录本,而是提议使用基础剪接过程的简化概率模型来量化可变剪接。我们的模型基于单个剪接位点的使用,并且可以生成任意复杂类型的剪接模式。在我们的实现 McSplicer 中,我们使用所有读取数据一次性估计模型的参数,我们在实验中证明,与竞争方法相比,这会产生更准确的估计。我们的模型能够使用少数易于解释的参数描述剪接突变的多种影响,正如我们在自闭症谱系障碍患者的 RNA-seq 数据实验中所说明的那样。
McSplicer 源代码可在 https://github.com/canzarlab/McSplicer 上获得,并已以存档格式存储在 https://doi.org/10.5281/zenodo.4449881 上。
补充数据可在《生物信息学》在线获得。