Patterson-Cross Ryan B, Levine Ariel J, Menon Vilas
Spinal Circuits and Plasticity Unit, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD, USA.
Department of Neurology, Center for Translational and Computational Neuroimmunology, Columbia University, New York City, NY, USA.
BMC Bioinformatics. 2021 Feb 1;22(1):39. doi: 10.1186/s12859-021-03957-4.
Generating and analysing single-cell data has become a widespread approach to examine tissue heterogeneity, and numerous algorithms exist for clustering these datasets to identify putative cell types with shared transcriptomic signatures. However, many of these clustering workflows rely on user-tuned parameter values, tailored to each dataset, to identify a set of biologically relevant clusters. Whereas users often develop their own intuition as to the optimal range of parameters for clustering on each data set, the lack of systematic approaches to identify this range can be daunting to new users of any given workflow. In addition, an optimal parameter set does not guarantee that all clusters are equally well-resolved, given the heterogeneity in transcriptomic signatures in most biological systems.
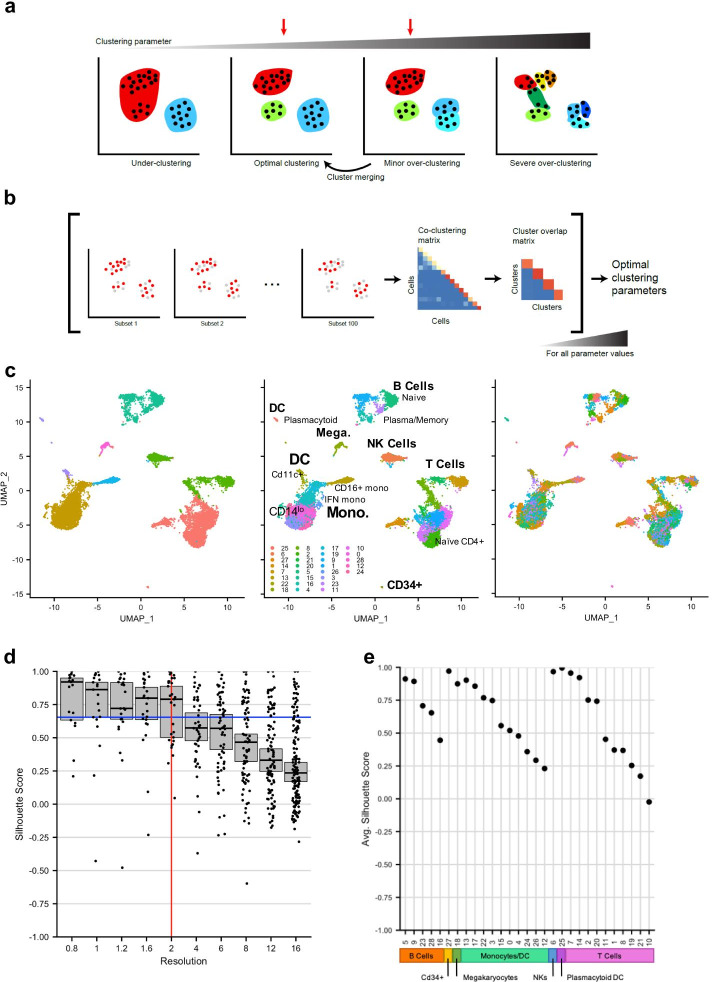
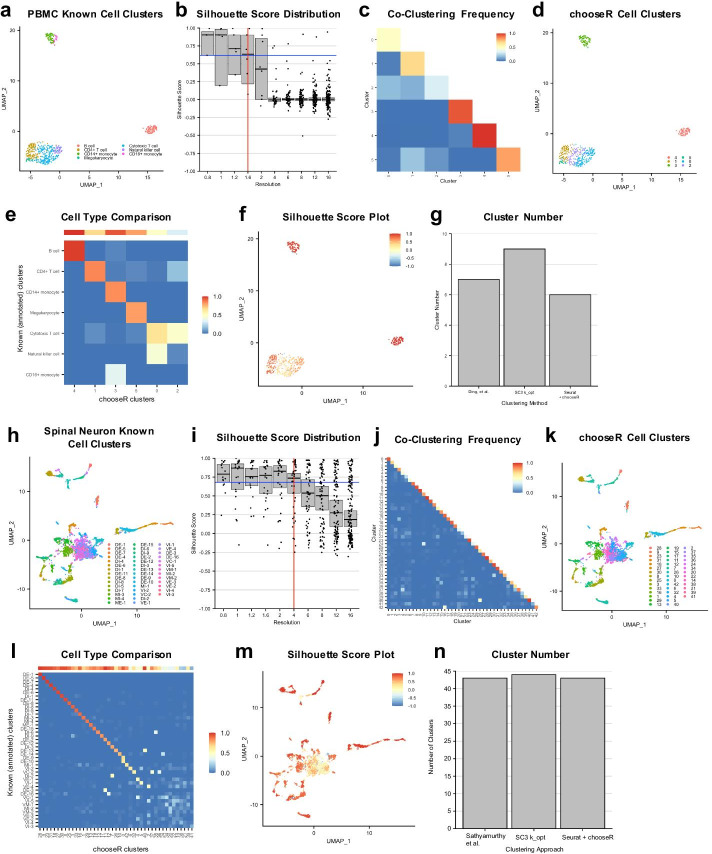
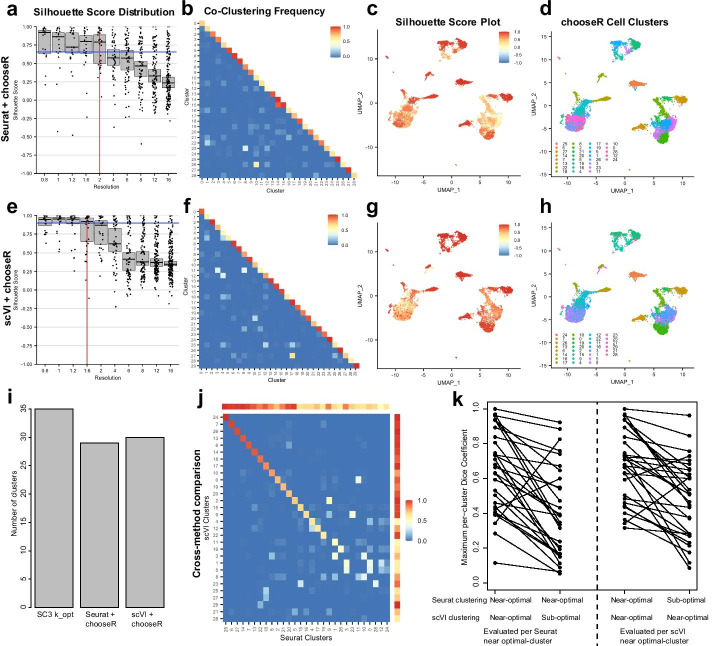
Here, we illustrate a subsampling-based approach (chooseR) that simultaneously guides parameter selection and characterizes cluster robustness. Through bootstrapped iterative clustering across a range of parameters, chooseR was used to select parameter values for two distinct clustering workflows (Seurat and scVI). In each case, chooseR identified parameters that produced biologically relevant clusters from both well-characterized (human PBMC) and complex (mouse spinal cord) datasets. Moreover, it provided a simple "robustness score" for each of these clusters, facilitating the assessment of cluster quality.
chooseR is a simple, conceptually understandable tool that can be used flexibly across clustering algorithms, workflows, and datasets to guide clustering parameter selection and characterize cluster robustness.
生成和分析单细胞数据已成为一种广泛应用的方法,用于研究组织异质性,并且存在许多算法可对这些数据集进行聚类,以识别具有共享转录组特征的假定细胞类型。然而,许多这些聚类工作流程依赖于针对每个数据集进行用户调整的参数值,以识别一组生物学相关的聚类。虽然用户通常会针对每个数据集的聚类参数最佳范围形成自己的直觉,但缺乏识别此范围的系统方法可能会让任何给定工作流程的新用户望而却步。此外,鉴于大多数生物系统中转录组特征的异质性,最优参数集并不能保证所有聚类都能得到同样好的解析。
在这里,我们展示了一种基于子采样的方法(chooseR),该方法可同时指导参数选择并表征聚类稳健性。通过在一系列参数上进行自举迭代聚类,chooseR被用于为两种不同的聚类工作流程(Seurat和scVI)选择参数值。在每种情况下,chooseR都能从特征明确的(人类外周血单核细胞)和复杂的(小鼠脊髓)数据集中识别出产生生物学相关聚类的参数。此外,它为每个聚类提供了一个简单的“稳健性分数”,便于评估聚类质量。
chooseR是一个简单、概念上易于理解的工具,可灵活应用于各种聚类算法、工作流程和数据集,以指导聚类参数选择并表征聚类稳健性。