Aishwarya S, Gunasekaran K, Sagaya Jansi R, Sangeetha G
Department of Bioinformatics, Stella Maris College (Autonomous), Chennai 600086, India.
Centre for Advanced Studies in Crystallography and Biophysics, University of Madras, Chennai 600025, India.
Comput Toxicol. 2021 May;18:100156. doi: 10.1016/j.comtox.2021.100156. Epub 2021 Jan 29.
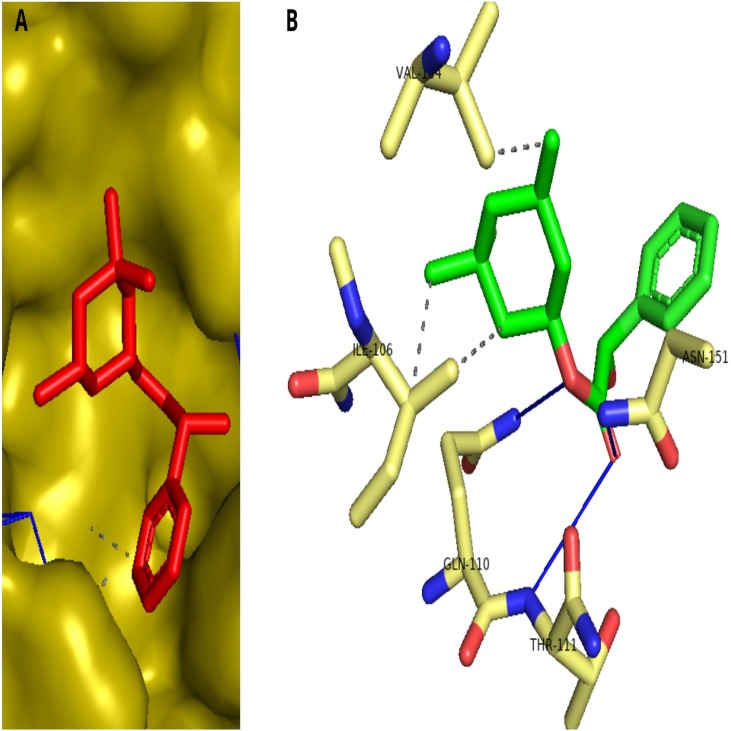
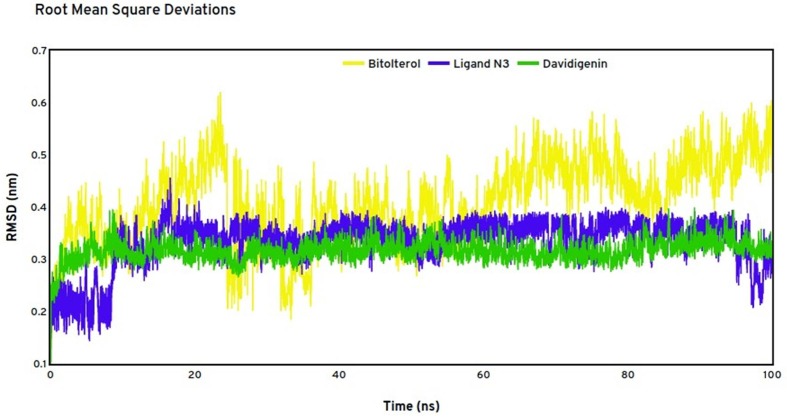
The recent pandemic Coronavirus disease-19 outbreak had traumatized global countries since its origin in late December 2019. Though the virus originated in China, it has spread rapidly across the world due its firmly established community transmission. To successfully tackle the spread and further infection, there needs a clear multidimensional understanding of the molecular mechanisms. Henceforth, 942 viral genome sequences were analysed to predict the core genomes crucial in virus life cycle. Additionally, 35 small interfering RNA transcripts were predicted that can target specifically the viral core proteins and reduce pathogenesis. The crystal structure of Covid-19 main protease-6LU7 was chosen as an attractive target due to the factors that there were fewer mutations and whose structure had significant identity to the annotated protein sequence of the core genome. Drug repurposing of both recruiting and non recruiting drugs was carried out through molecular docking procedures to recognize bitolterol as a good inhibitor of Covid-19 protease. The study was extended further to screen antiviral phytocompounds through quantitative structure activity relationship and molecular docking to identify davidigenin, from licorice as the best novel lead with good interactions and binding energy. The docking of the best compounds in all three categories was validated with molecular dynamics simulations which implied stable binding of the drug and lead molecule. Though the studies need clinical evaluations, the results are suggestive of curbing the pandemic.
自2019年12月底起源以来,近期的新冠疫情已使全球各国遭受重创。尽管该病毒起源于中国,但由于其已牢固确立的社区传播,它已在全球迅速传播。为了成功应对病毒传播及进一步感染,需要对分子机制有清晰的多维度理解。因此,分析了942个病毒基因组序列,以预测在病毒生命周期中至关重要的核心基因组。此外,还预测了35个小干扰RNA转录本,它们可以特异性靶向病毒核心蛋白并降低致病性。新冠病毒主要蛋白酶-6LU7的晶体结构因其突变较少且其结构与核心基因组的注释蛋白序列具有显著同源性而被选为有吸引力的靶点。通过分子对接程序对招募性和非招募性药物进行药物再利用,以识别比托特罗为新冠病毒蛋白酶的良好抑制剂。该研究进一步扩展,通过定量构效关系和分子对接筛选抗病毒植物化合物,以确定甘草中的光甘草定作为具有良好相互作用和结合能的最佳新型先导物。所有三类最佳化合物的对接都通过分子动力学模拟进行了验证,这表明药物和先导分子的结合稳定。尽管这些研究需要临床评估,但结果表明有可能遏制疫情。