He Zongzhen, Zhang Junying, Yuan Xiguo, Zhang Yuanyuan
School of Computer Science and Technology, Xidian University, Xi'an, China.
School of Information and Control Engineering, Qingdao University of Technology, Qingdao, China.
Front Genet. 2021 Jan 18;11:632901. doi: 10.3389/fgene.2020.632901. eCollection 2020.
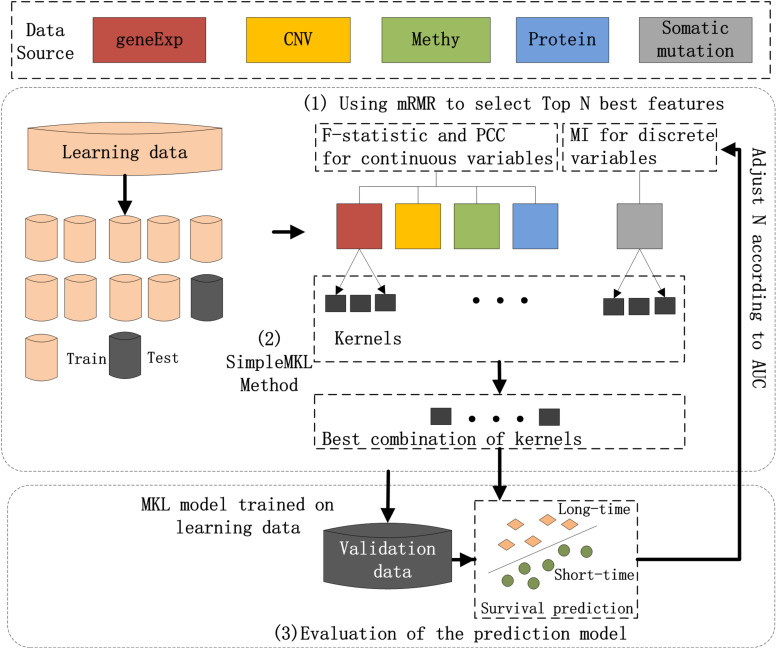
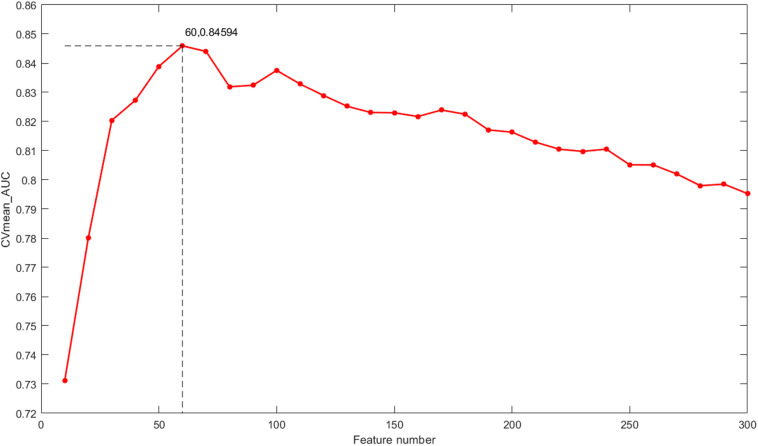
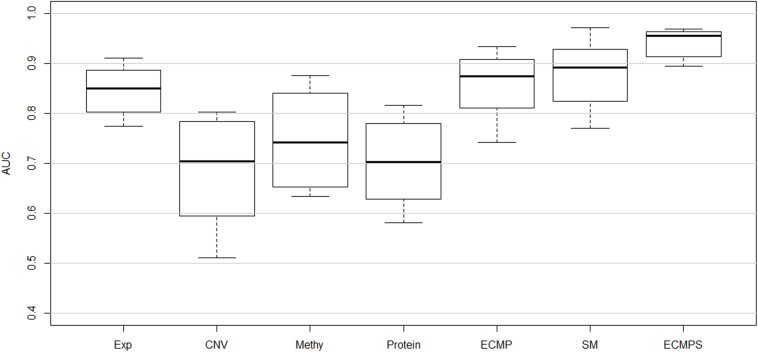
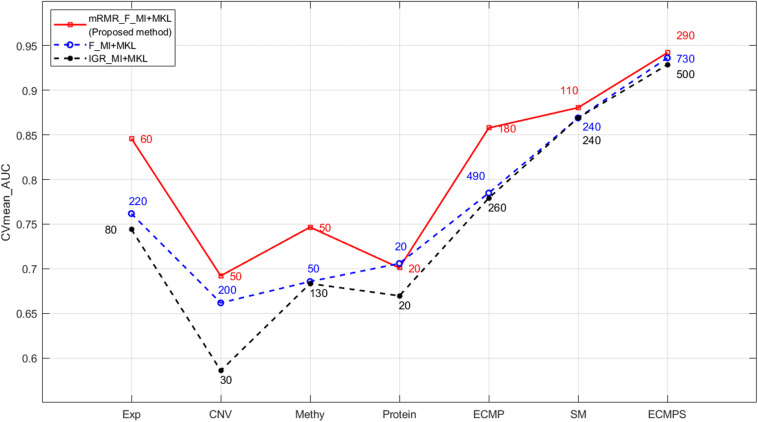
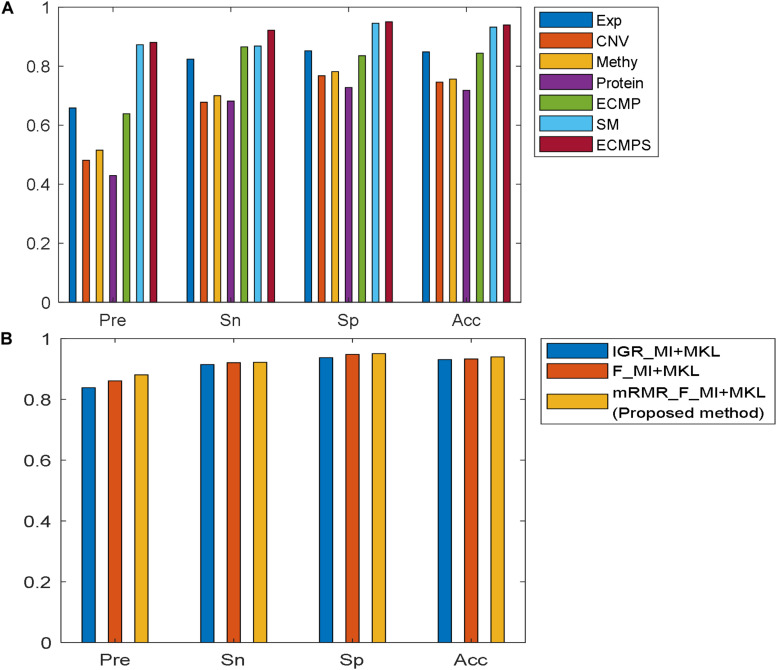
Breast cancer is the most common malignancy in women, and because it has a high mortality rate, it is urgent to develop computational methods to increase the accuracy of breast cancer survival predictive models. Although multi-omics data such as gene expression have been extensively used in recent studies, the accurate prognosis of breast cancer remains a challenge. Somatic mutations are another important and promising data source for studying cancer development, and its effect on the prognosis of breast cancer remains to be further explored. Meanwhile, these omics datasets are high-dimensional and redundant. Therefore, we adopted multiple kernel learning (MKL) to efficiently integrate somatic mutation to currently molecular data including gene expression, copy number variation (CNV), methylation, and protein expression data for the prediction of breast cancer survival. Before integration, the maximum relevance minimum redundancy (mRMR) feature selection method was utilized to select features that present high relevance to survival and low redundancy among themselves for each type of data. The experimental results demonstrated that the proposed method achieved the most optimal performance and there was a remarkable improvement in the prediction performance when somatic mutations were included, indicating that somatic mutations are critical for improving breast cancer survival predictions. Moreover, mRMR was superior to other feature selection methods used in previous studies. Furthermore, MKL outperformed the other traditional classifiers in multi-omics data integration. Our analysis indicated that through employing promising omics data such as somatic mutations and harnessing the power of proper feature selection methods and effective integration frameworks, the breast cancer survival predictive accuracy can be further increased, thereby providing a more optimal clinical diagnosis and more effective treatment for breast cancer patients.
乳腺癌是女性中最常见的恶性肿瘤,由于其死亡率高,因此迫切需要开发计算方法以提高乳腺癌生存预测模型的准确性。尽管诸如基因表达等多组学数据在最近的研究中已被广泛使用,但乳腺癌的准确预后仍然是一个挑战。体细胞突变是研究癌症发展的另一个重要且有前景的数据来源,其对乳腺癌预后的影响仍有待进一步探索。同时,这些组学数据集具有高维度和冗余性。因此,我们采用多核学习(MKL)来有效地将体细胞突变与当前的分子数据(包括基因表达、拷贝数变异(CNV)、甲基化和蛋白质表达数据)整合起来,以预测乳腺癌的生存情况。在整合之前,利用最大相关最小冗余(mRMR)特征选择方法为每种类型的数据选择与生存高度相关且彼此之间冗余度低的特征。实验结果表明,该方法实现了最优性能,并且纳入体细胞突变后预测性能有显著提高,这表明体细胞突变对于改善乳腺癌生存预测至关重要。此外,mRMR优于先前研究中使用的其他特征选择方法。此外,在多组学数据整合方面,MKL优于其他传统分类器。我们的分析表明,通过采用体细胞突变等有前景的组学数据,并利用适当的特征选择方法和有效的整合框架,可以进一步提高乳腺癌生存预测的准确性,从而为乳腺癌患者提供更优化的临床诊断和更有效的治疗。