Bovio-Winkler Patricia, Cabezas Angela, Etchebehere Claudia
Microbial Ecology Laboratory, Department of Microbial Biochemistry and Genomic, Biological Research Institute "Clemente Estable," Montevideo, Uruguay.
Instituto Tecnológico Regional Centro Sur, Universidad Tecnológica, Durazno, Uruguay.
Front Microbiol. 2021 Jan 20;11:603234. doi: 10.3389/fmicb.2020.603234. eCollection 2020.
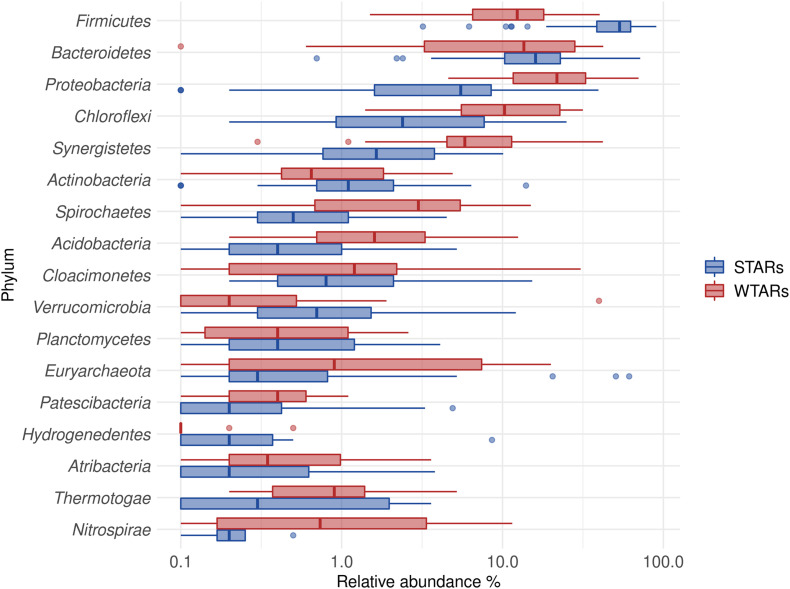
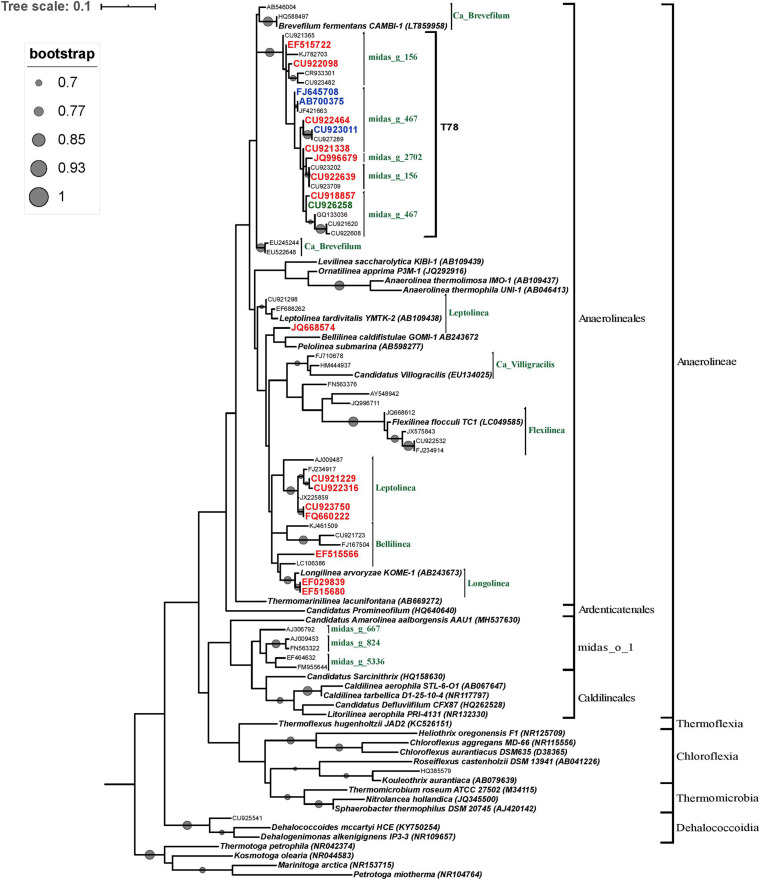
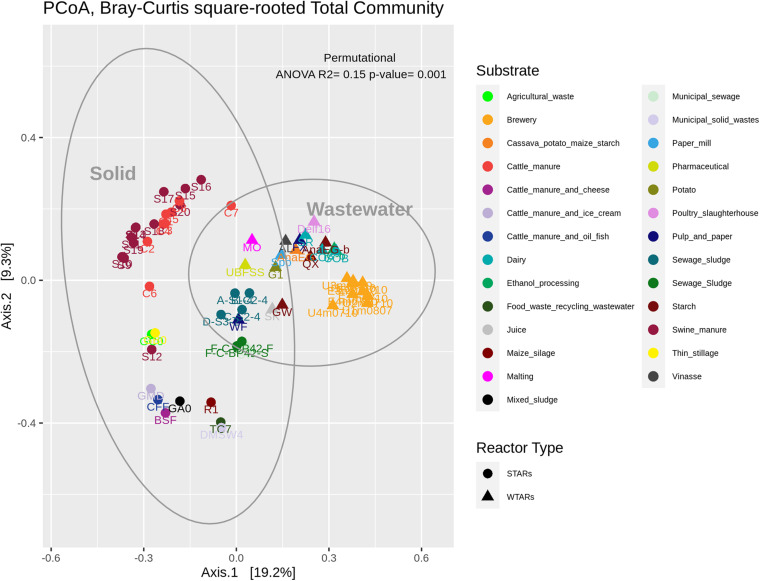
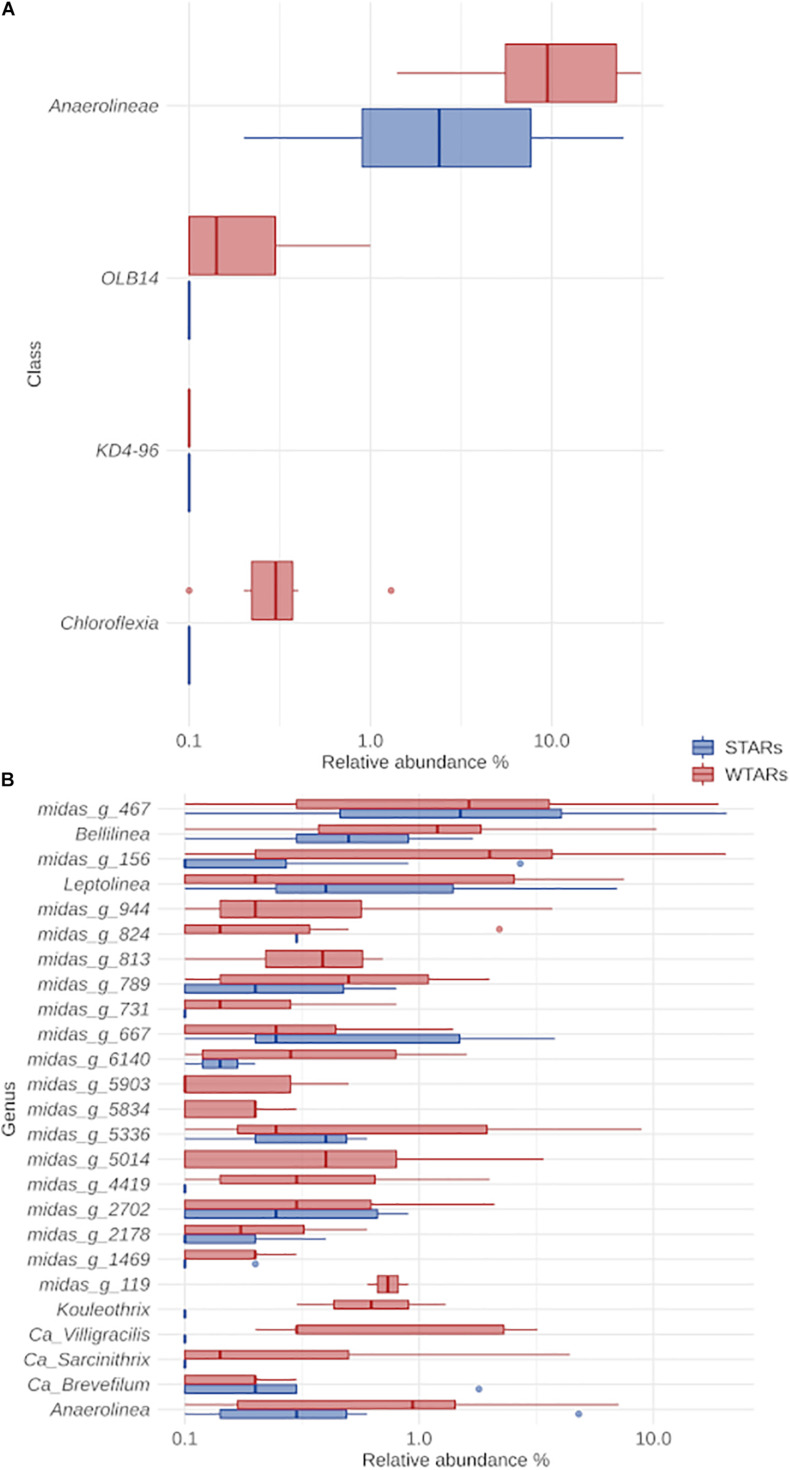
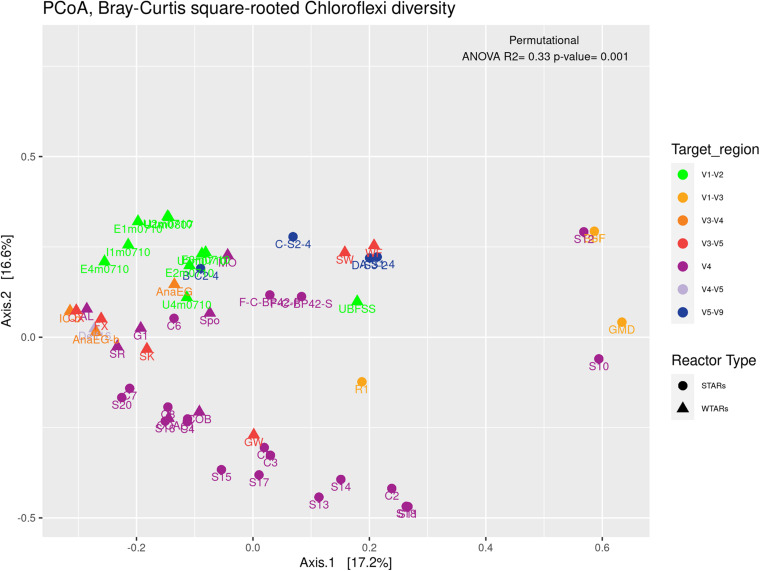
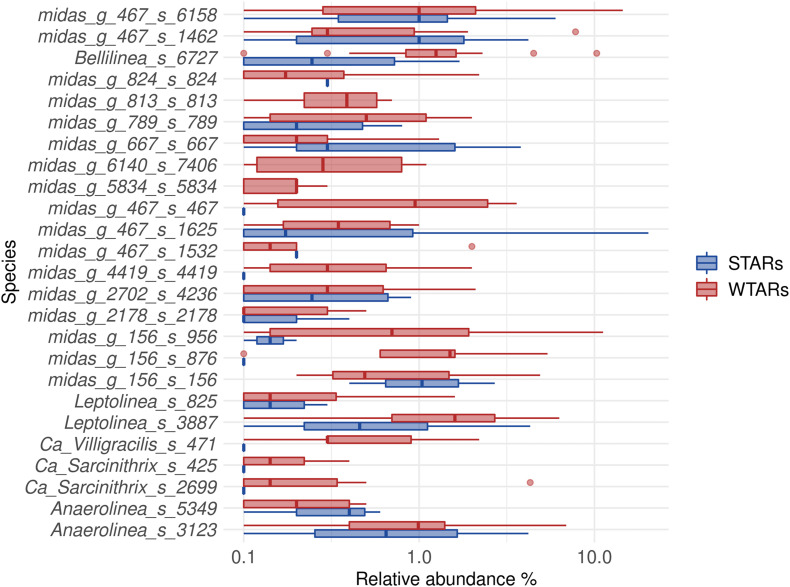
Although microbial communities of anaerobic bioreactors have been extensively studied using DNA-based tools, there are still several knowledge gaps regarding the microbiology of the process, in particular integration of all generated data is still limited. One understudied core phylum within anaerobic bioreactors is the phylum Chloroflexi, despite being one of the most abundant groups in anaerobic reactors. In order to address the abundance, diversity and phylogeny of this group in full-scale methanogenic reactors globally distributed, a compilation of 16S ribosomal RNA gene sequence data from 62 full-scale methanogenic reactors studied worldwide, fed either with wastewater treatment anaerobic reactors (WTARs) or solid-waste treatment anaerobic reactors (STARs), was performed. One of the barriers to overcome was comparing data generated using different primer sets and different sequencing platforms. The sequence analysis revealed that the average abundance of Chloroflexi in WTARs was higher than in STARs. Four genera belonging to the Anaerolineae class dominated both WTARs and STARs but the core populations were different. According to the phylogenetic analysis, most of the sequences formed clusters with no cultured representatives. The Anaerolineae class was more abundant in reactors with granular biomass than in reactors with disperse biomass supporting the hypothesis that Anaerolineae play an important role in granule formation and structure due to their filamentous morphology. Cross-study comparisons can be fruitfully used to understand the complexity of the anaerobic digestion process. However, more efforts are needed to standardize protocols and report metadata information.
尽管已使用基于DNA的工具对厌氧生物反应器的微生物群落进行了广泛研究,但在该过程的微生物学方面仍存在一些知识空白,特别是所有生成数据的整合仍然有限。厌氧生物反应器中一个研究较少的核心门类是绿弯菌门,尽管它是厌氧反应器中最丰富的菌群之一。为了全面了解全球分布的全尺寸产甲烷反应器中该菌群的丰度、多样性和系统发育,我们对来自全球62个全尺寸产甲烷反应器的16S核糖体RNA基因序列数据进行了汇编,这些反应器以废水处理厌氧反应器(WTAR)或固体废物处理厌氧反应器(STAR)为进料。需要克服的障碍之一是比较使用不同引物组和不同测序平台生成的数据。序列分析表明,WTAR中绿弯菌门的平均丰度高于STAR。属于厌氧绳菌纲的四个属在WTAR和STAR中均占主导地位,但核心种群不同。根据系统发育分析,大多数序列形成了没有培养代表的簇。厌氧绳菌纲在具有颗粒状生物质的反应器中比在具有分散生物质的反应器中更为丰富,这支持了厌氧绳菌纲因其丝状形态在颗粒形成和结构中起重要作用的假设。跨研究比较可有效地用于理解厌氧消化过程的复杂性。然而,需要更多努力来规范方案并报告元数据信息。