University of Natural Resources and Life Sciences (BOKU), Vienna, Austria.
São Paulo State University (Unesp), School of Veterinary Medicine, Department of Production and Animal Health, Araçatuba, São Paulo, Brazil.
BMC Genomics. 2021 Feb 8;22(1):108. doi: 10.1186/s12864-021-07412-9.
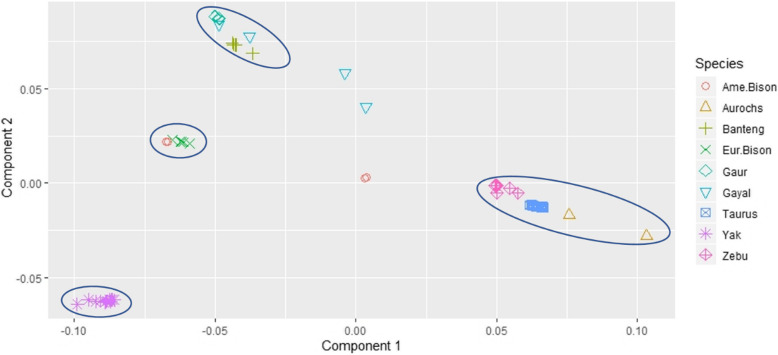
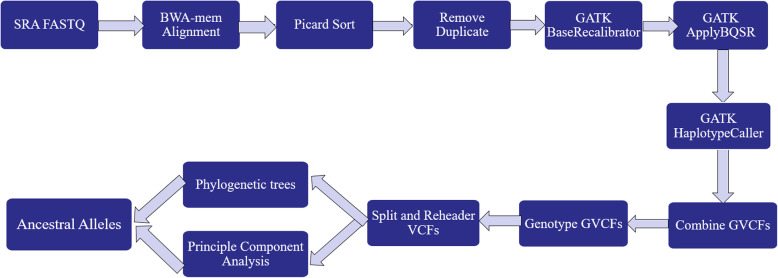
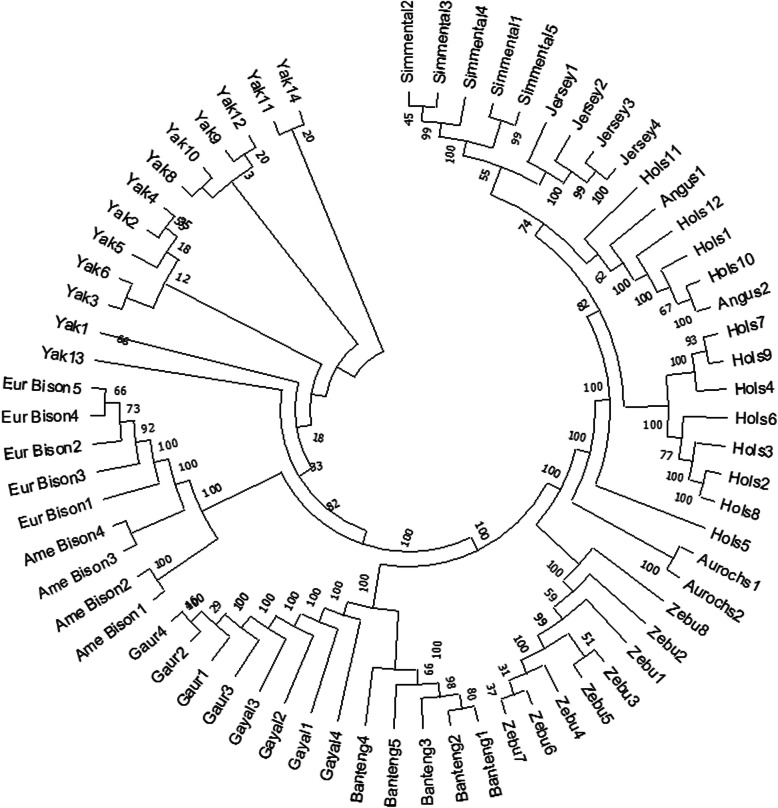
In evolutionary theory, divergence and speciation can arise from long periods of reproductive isolation, genetic mutation, selection and environmental adaptation. After divergence, alleles can either persist in their initial state (ancestral allele - AA), co-exist or be replaced by a mutated state (derived alleles -DA). In this study, we aligned whole genome sequences of individuals from the Bovinae subfamily to the cattle reference genome (ARS.UCD-1.2) for defining ancestral alleles necessary for selection signatures study.
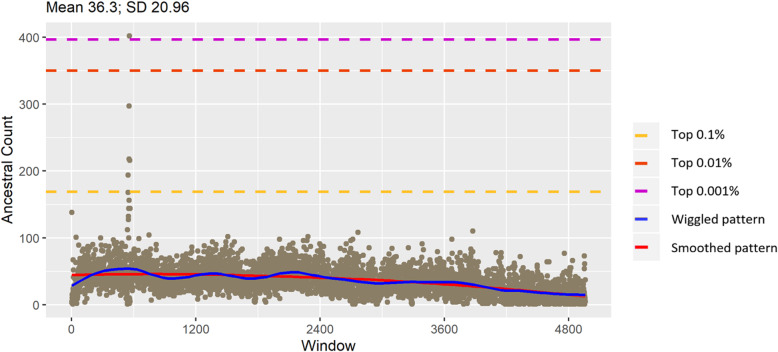
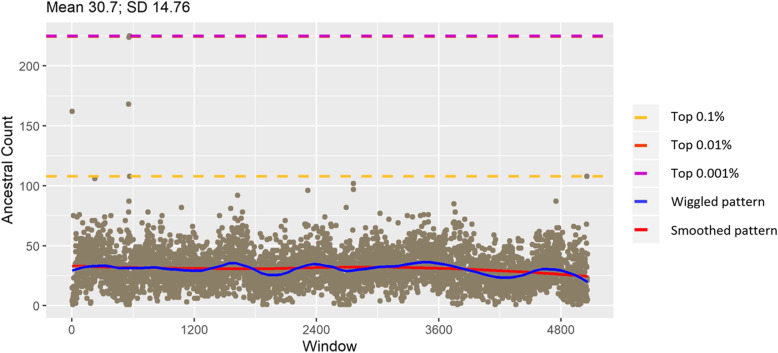
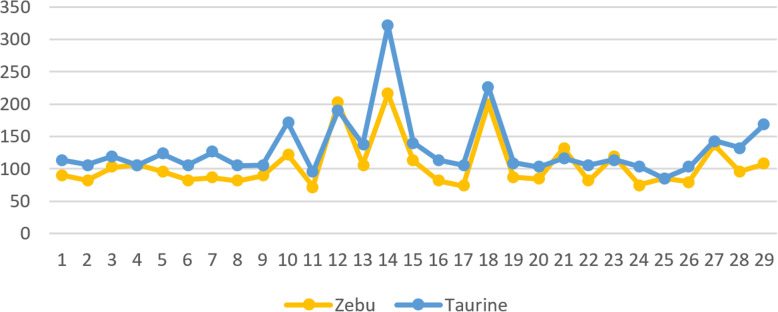
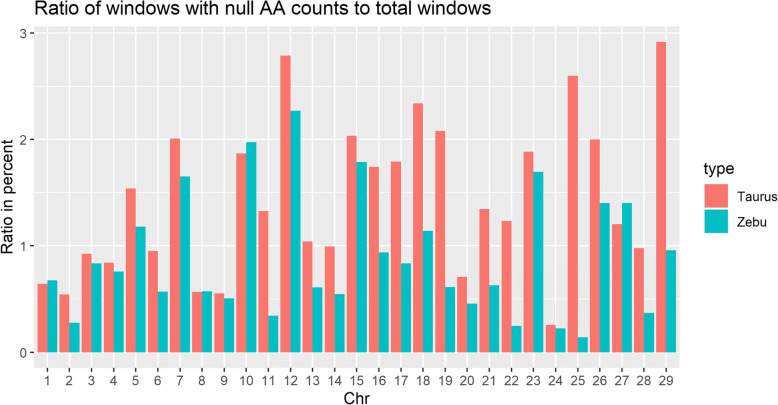
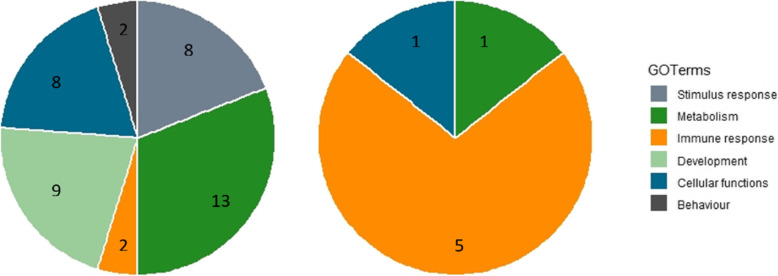
Accommodating independent divergent of each lineage from the initial ancestral state, AA were defined based on fixed alleles on at least two groups of yak, bison and gayal-gaur-banteng resulting in ~ 32.4 million variants. Using non-overlapping scanning windows of 10 Kb, we counted the AA observed within taurine and zebu cattle. We focused on the extreme points, regions with top 0. 1% (high count) and regions without any occurrence of AA (null count). High count regions preserved gene functions from ancestral states that are still beneficial in the current condition, while null counts regions were linked to mutated ones. For both cattle, high count regions were associated with basal lipid metabolism, essential for survival of various environmental pressures. Mutated regions were associated to productive traits in taurine, i.e. higher metabolism, cell development and behaviors and in immune response domain for zebu.
Our findings suggest that retaining and losing AA in some regions are varied and made it species-specific with possibility of overlapping as it depends on the selective pressure they had to experience.
在进化理论中,分歧和物种形成可能源于长时间的生殖隔离、基因突变、选择和环境适应。分歧后,等位基因可以保持在初始状态(祖先等位基因 - AA),共存或被突变状态取代(衍生等位基因 - DA)。在这项研究中,我们将牛亚科个体的全基因组序列与牛参考基因组(ARS.UCD-1.2)进行比对,以确定用于选择特征研究的祖先等位基因。
为了适应每个谱系从初始祖先状态的独立分歧,我们根据至少两组牦牛、野牛和野牦牛-野牛-大额牛的固定等位基因来定义 AA,导致约 3240 万个变体。使用非重叠的 10 Kb 扫描窗口,我们计算了在黄牛和瘤牛中观察到的 AA。我们关注极端点、具有前 0.1%(高计数)的区域和没有任何 AA 出现的区域(零计数)。高计数区域保留了祖先状态的基因功能,这些功能在当前条件下仍然是有益的,而零计数区域则与突变有关。对于这两种牛,高计数区域与基础脂质代谢有关,这对各种环境压力下的生存至关重要。突变区域与黄牛的生产性状有关,即更高的新陈代谢、细胞发育和行为,以及瘤牛的免疫反应域。
我们的发现表明,在某些区域保留和丢失 AA 是多样化的,并且因物种而异,可能存在重叠,这取决于它们必须经历的选择压力。