Faculty of Veterinary and Agricultural Science, The University of Melbourne, Parkville, VIC, Australia.
Agriculture Victoria, AgriBio, Centre for AgriBiosciences, Bundoora, VIC, Australia.
Nat Commun. 2021 Feb 8;12(1):860. doi: 10.1038/s41467-021-21001-0.
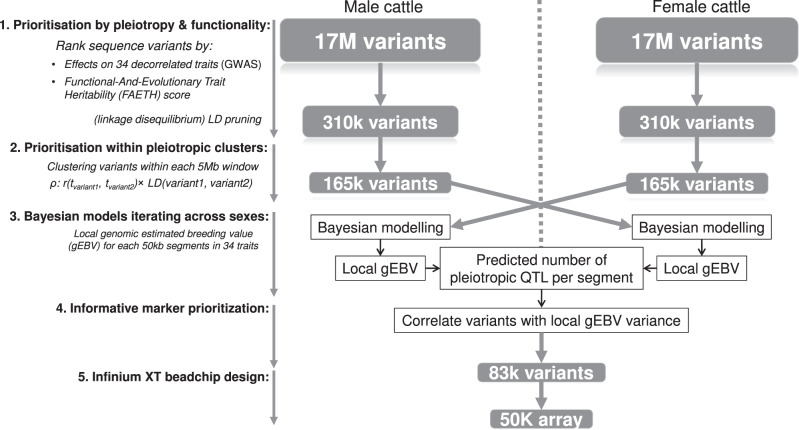
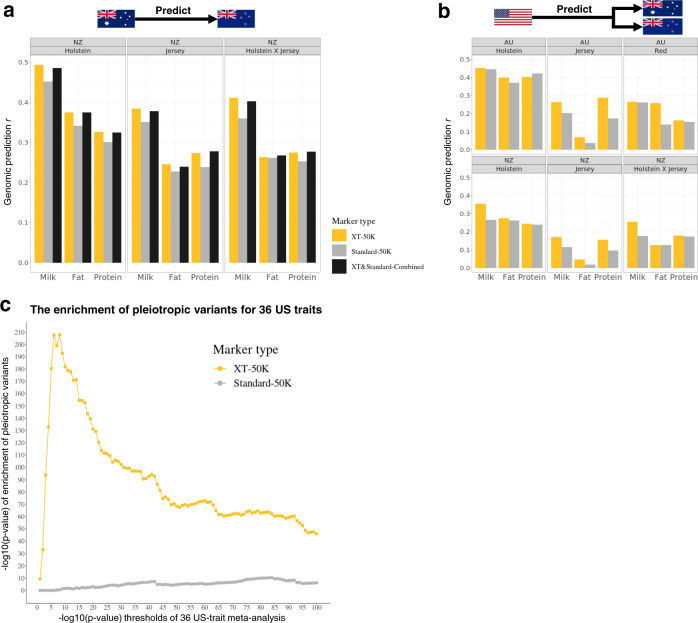
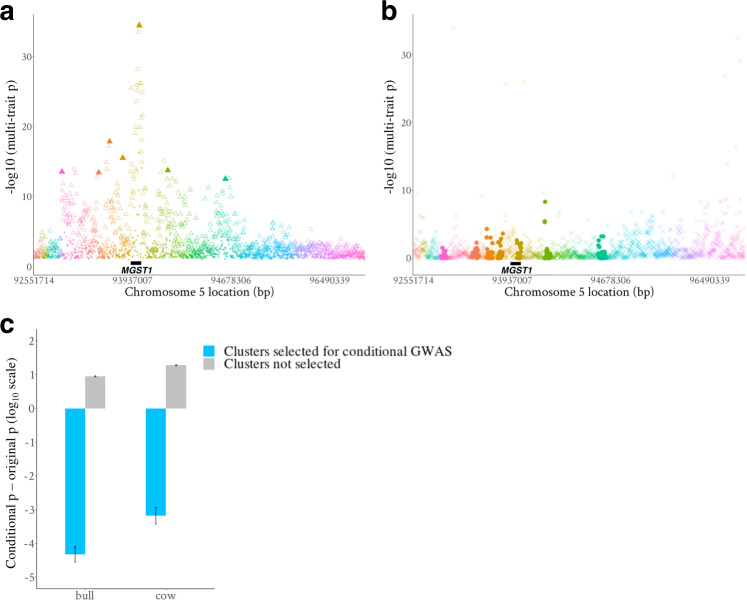
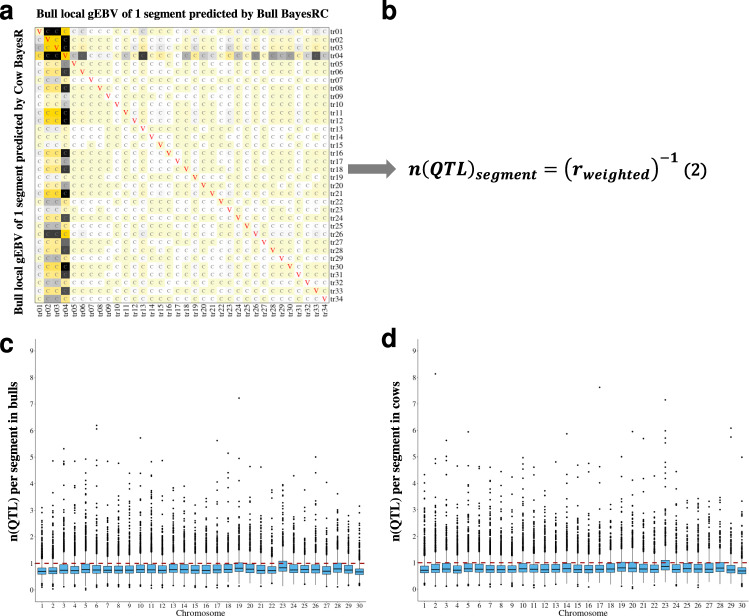
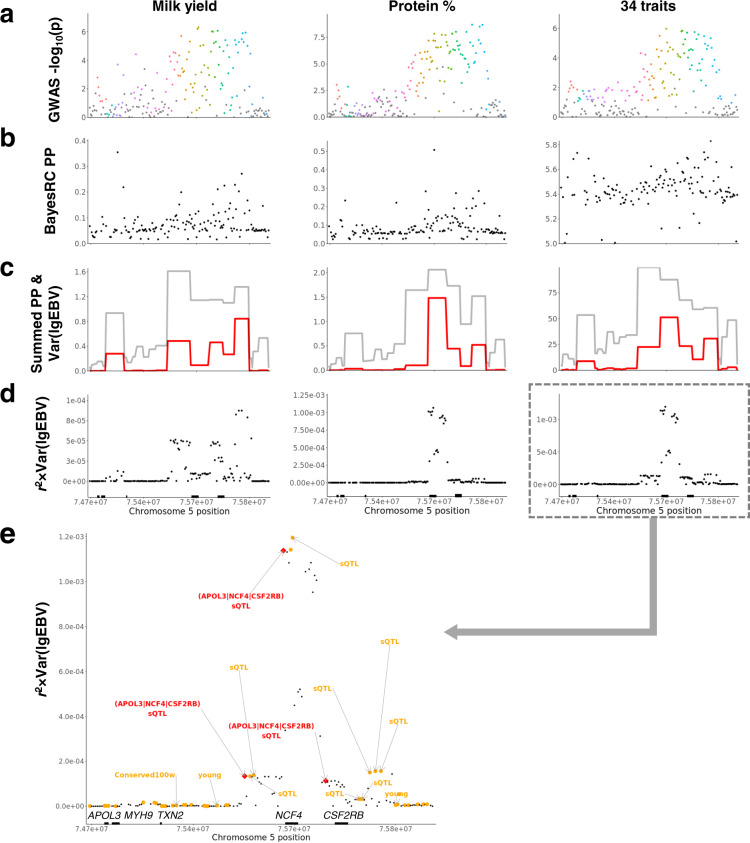
The difficulty in finding causative mutations has hampered their use in genomic prediction. Here, we present a methodology to fine-map potentially causal variants genome-wide by integrating the functional, evolutionary and pleiotropic information of variants using GWAS, variant clustering and Bayesian mixture models. Our analysis of 17 million sequence variants in 44,000+ Australian dairy cattle for 34 traits suggests, on average, one pleiotropic QTL existing in each 50 kb chromosome-segment. We selected a set of 80k variants representing potentially causal variants within each chromosome segment to develop a bovine XT-50K genotyping array. The custom array contains many pleiotropic variants with biological functions, including splicing QTLs and variants at conserved sites across 100 vertebrate species. This biology-informed custom array outperformed the standard array in predicting genetic value of multiple traits across populations in independent datasets of 90,000+ dairy cattle from the USA, Australia and New Zealand.
发现致病突变的困难阻碍了它们在基因组预测中的应用。在这里,我们提出了一种通过整合 GWAS、变异聚类和贝叶斯混合模型来对全基因组潜在因果变异进行精细映射的方法。我们对 44000 多头澳大利亚奶牛的 1700 万个序列变异进行了分析,研究了 34 个特征,结果表明,平均每个 50kb 的染色体片段存在一个多效性 QTL。我们选择了一组代表每个染色体片段中潜在因果变异的 80k 个变体,以开发牛 XT-50K 基因分型阵列。定制阵列包含许多具有生物学功能的多效性变体,包括剪接 QTL 和在 100 种脊椎动物物种中保守位点的变体。这种基于生物学的定制阵列在预测来自美国、澳大利亚和新西兰的 90000 多头奶牛的多个群体的遗传值方面优于标准阵列。