D. E. Shaw Research, New York, NY, 10036, USA.
Department of Biochemistry and Molecular Biophysics, Columbia University, New York, NY, 10032, USA.
Sci Data. 2021 Feb 10;8(1):55. doi: 10.1038/s41597-021-00833-x.
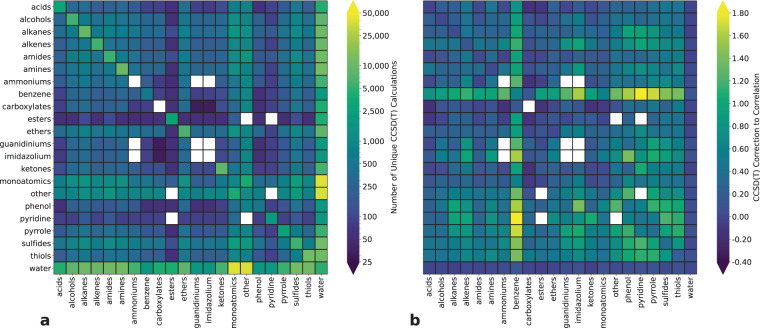
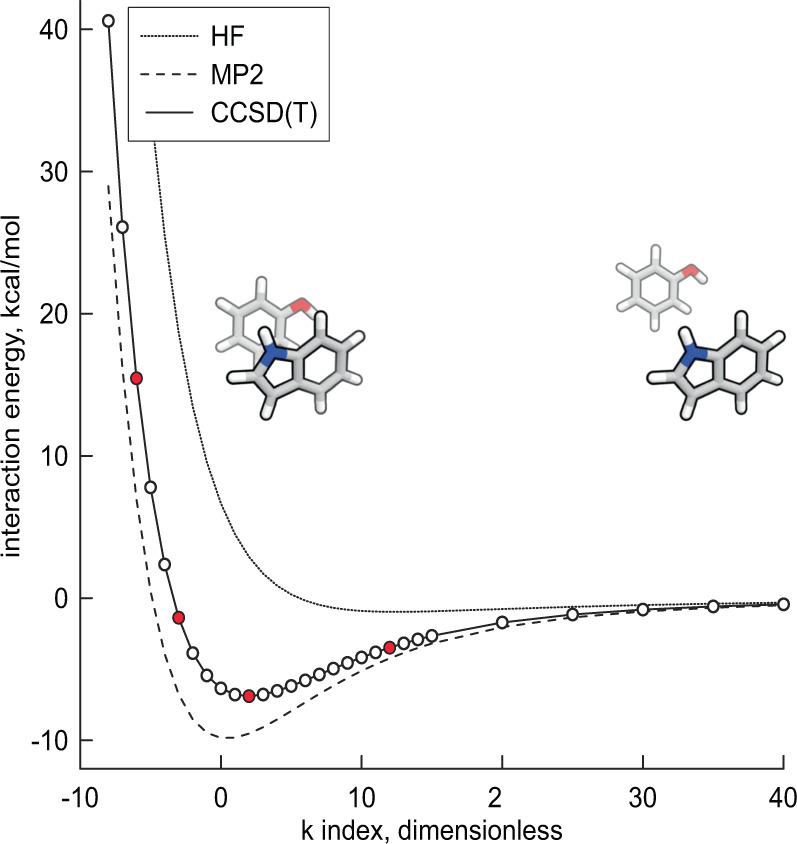
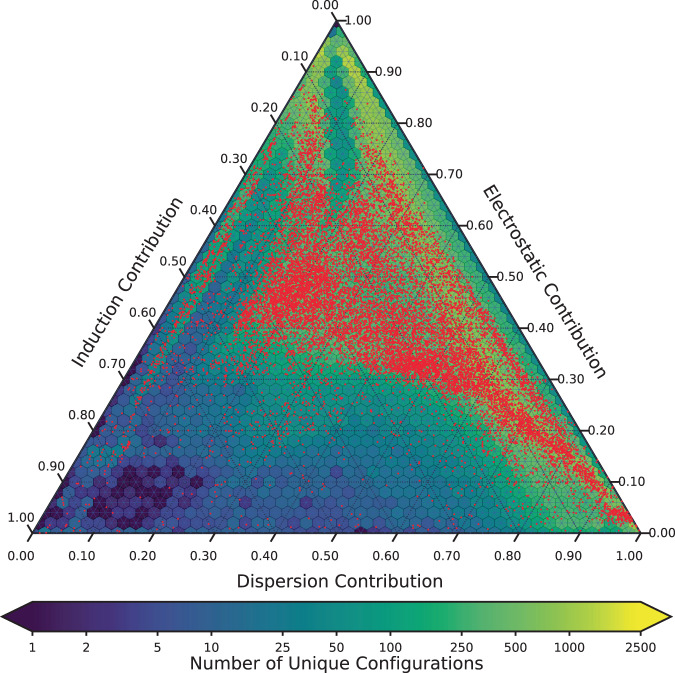
Advances in computational chemistry create an ongoing need for larger and higher-quality datasets that characterize noncovalent molecular interactions. We present three benchmark collections of quantum mechanical data, covering approximately 3,700 distinct types of interacting molecule pairs. The first collection, which we refer to as DES370K, contains interaction energies for more than 370,000 dimer geometries. These were computed using the coupled-cluster method with single, double, and perturbative triple excitations [CCSD(T)], which is widely regarded as the gold-standard method in electronic structure theory. Our second benchmark collection, a core representative subset of DES370K called DES15K, is intended for more computationally demanding applications of the data. Finally, DES5M, our third collection, comprises interaction energies for nearly 5,000,000 dimer geometries; these were calculated using SNS-MP2, a machine learning approach that provides results with accuracy comparable to that of our coupled-cluster training data. These datasets may prove useful in the development of density functionals, empirically corrected wavefunction-based approaches, semi-empirical methods, force fields, and models trained using machine learning methods.
计算化学的进展不断需要更大和更高质量的数据集来描述非共价分子相互作用。我们提出了三个量子力学数据集的基准集,涵盖了大约 3700 种不同类型的相互作用分子对。第一个数据集,我们称之为 DES370K,包含了超过 370000 个二聚体结构的相互作用能。这些是使用包含单、双和微扰三重激发的耦合簇方法[CCSD(T)]计算得到的,该方法被广泛认为是电子结构理论中的黄金标准方法。我们的第二个基准数据集,DES370K 的一个核心代表子集,称为 DES15K,是为了更具挑战性的数据集应用而设计的。最后,我们的第三个数据集 DES5M,包含了近 5000000 个二聚体结构的相互作用能,这些是使用 SNS-MP2 计算得到的,SNS-MP2 是一种机器学习方法,其结果的准确性与我们的耦合簇训练数据相当。这些数据集可能在密度泛函、经验修正波函数方法、半经验方法、力场和使用机器学习方法训练的模型的开发中证明是有用的。