Institute for Molecular Bioscience, The University of Queensland, Brisbane, QLD, Australia.
Faculty of Veterinary and Agricultural Science, University of Melbourne, Parkville, VIC, Australia.
Nat Commun. 2021 Feb 19;12(1):1164. doi: 10.1038/s41467-021-21446-3.
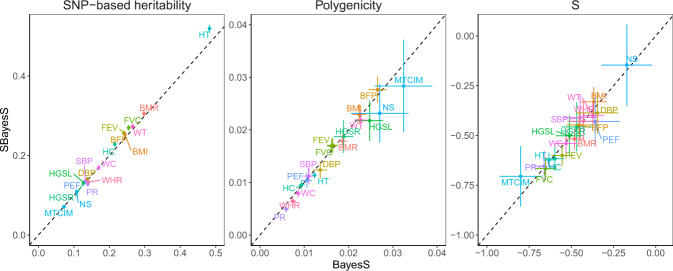
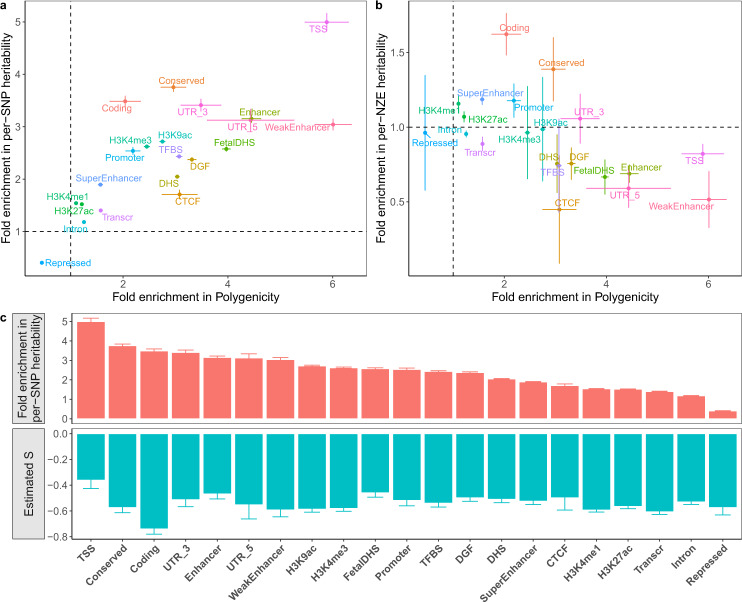
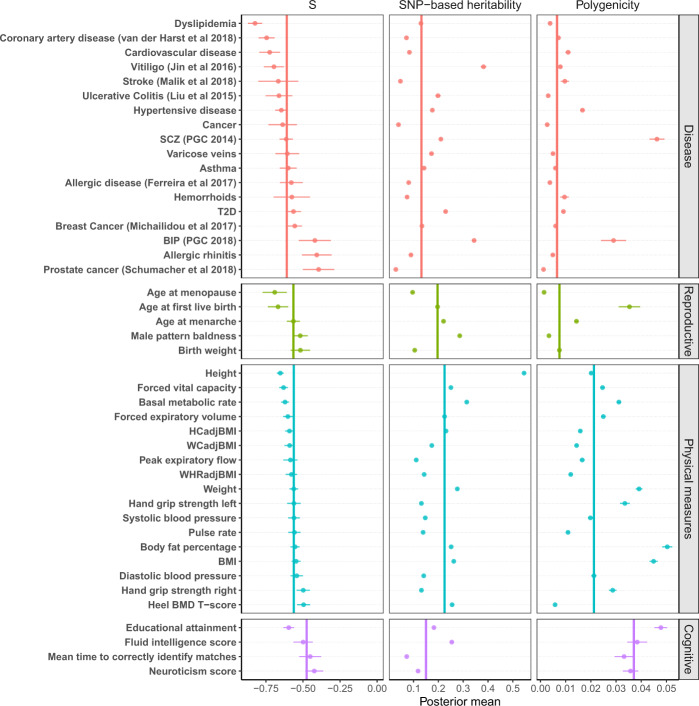
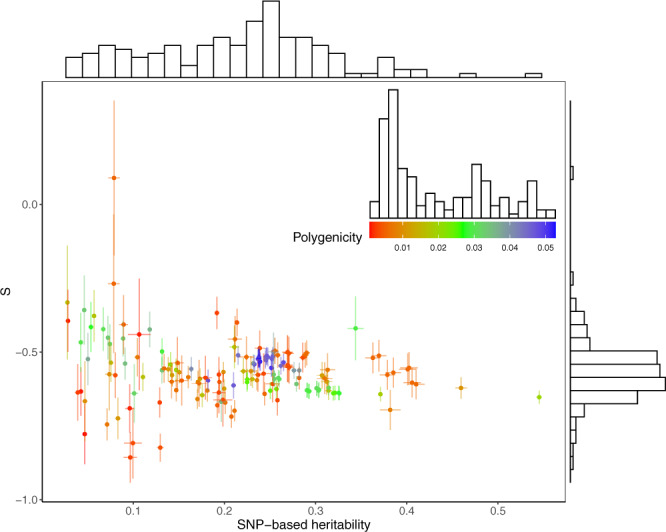
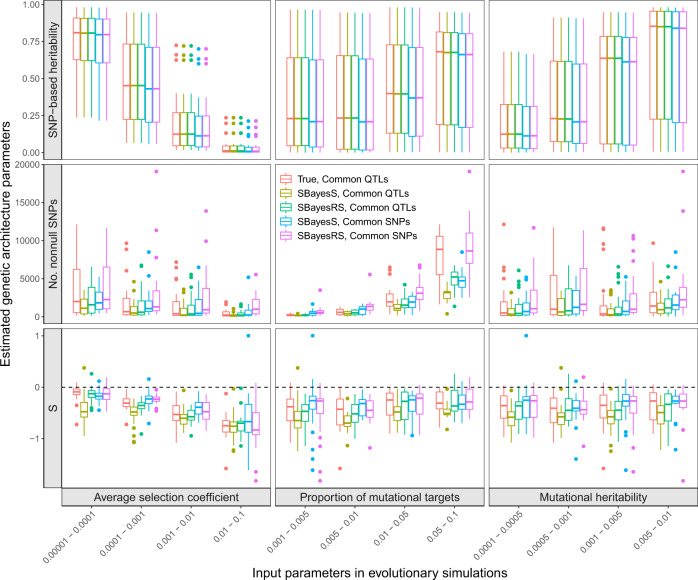
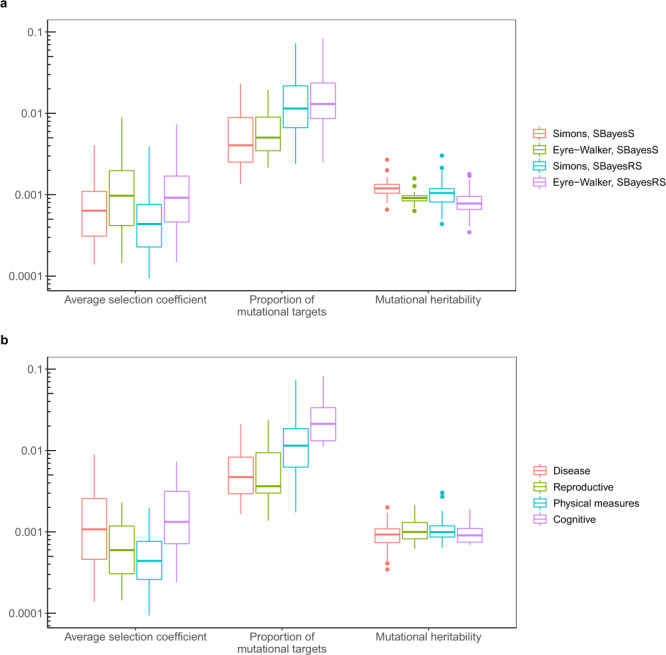
Understanding how natural selection has shaped genetic architecture of complex traits is of importance in medical and evolutionary genetics. Bayesian methods have been developed using individual-level GWAS data to estimate multiple genetic architecture parameters including selection signature. Here, we present a method (SBayesS) that only requires GWAS summary statistics. We analyse data for 155 complex traits (n = 27k-547k) and project the estimates onto those obtained from evolutionary simulations. We estimate that, on average across traits, about 1% of human genome sequence are mutational targets with a mean selection coefficient of ~0.001. Common diseases, on average, show a smaller number of mutational targets and have been under stronger selection, compared to other traits. SBayesS analyses incorporating functional annotations reveal that selection signatures vary across genomic regions, among which coding regions have the strongest selection signature and are enriched for both the number of associated variants and the magnitude of effect sizes.
了解自然选择如何塑造复杂性状的遗传结构对于医学和进化遗传学都很重要。贝叶斯方法已经被开发出来,使用个体水平的 GWAS 数据来估计多个遗传结构参数,包括选择特征。在这里,我们提出了一种仅需要 GWAS 汇总统计数据的方法(SBayesS)。我们分析了 155 种复杂性状的数据(n = 27k-547k),并将这些估计值投射到从进化模拟中获得的估计值上。我们估计,在平均每个性状中,大约有 1%的人类基因组序列是突变靶标,平均选择系数约为 0.001。与其他性状相比,常见疾病的突变靶标数量较少,受到的选择压力也更强。纳入功能注释的 SBayesS 分析表明,选择特征在基因组区域之间存在差异,其中编码区域的选择特征最强,并且与相关变异数量和效应大小幅度都富集。