CY Advanced Studies and LPTM (UMR8089 of CNRS), CY Cergy-Paris Université, 95302, Cergy-Pontoise, France.
Department of Physics, University of Warwick, Coventry, CV4 7AL, UK.
Sci Rep. 2021 Feb 19;11(1):4257. doi: 10.1038/s41598-021-82849-2.
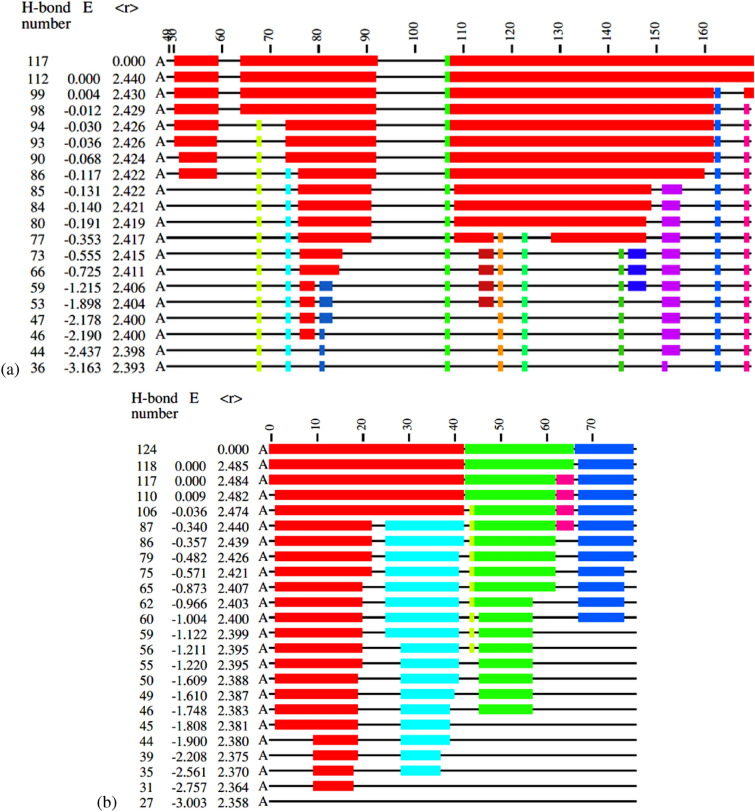
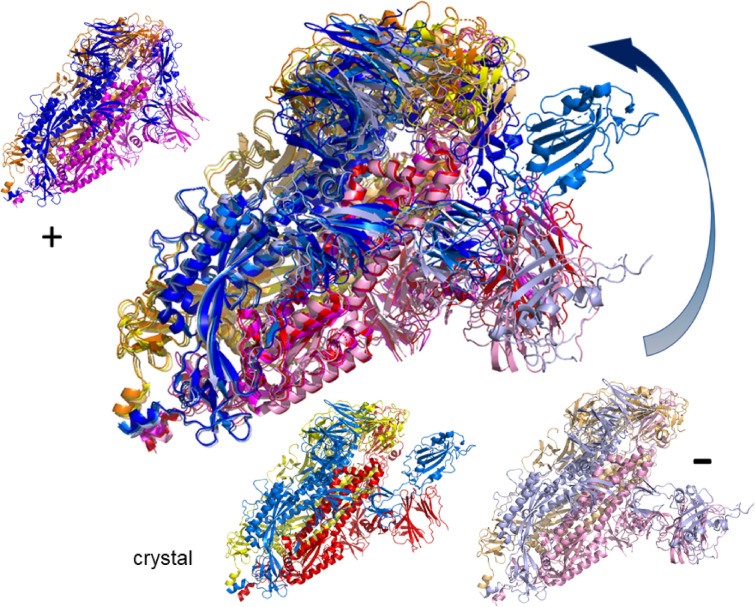
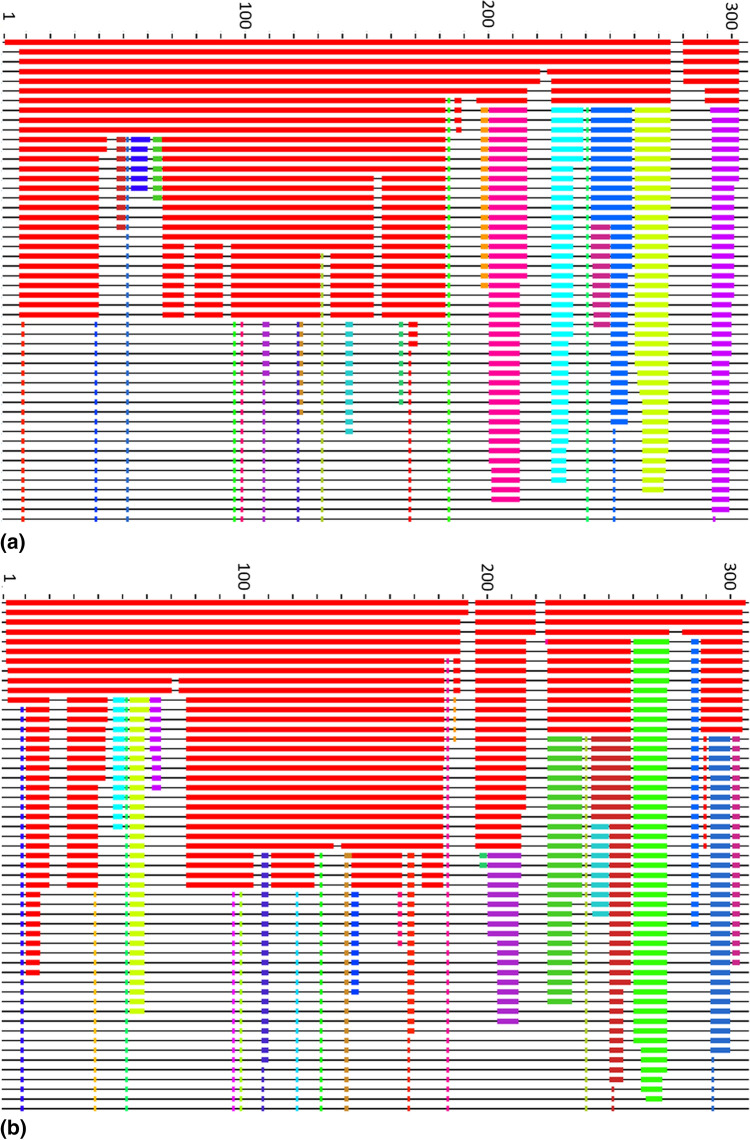
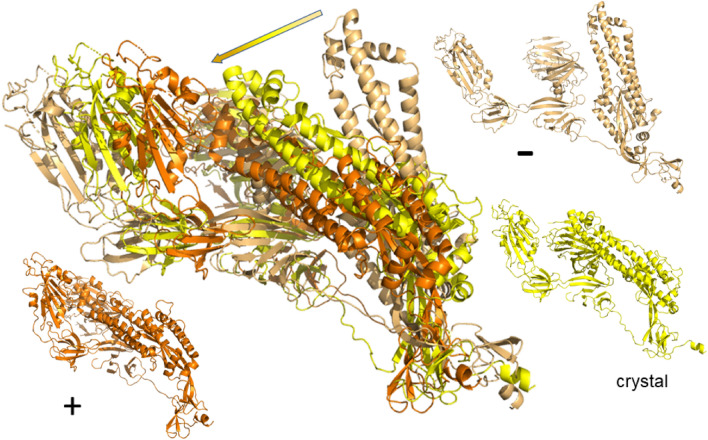
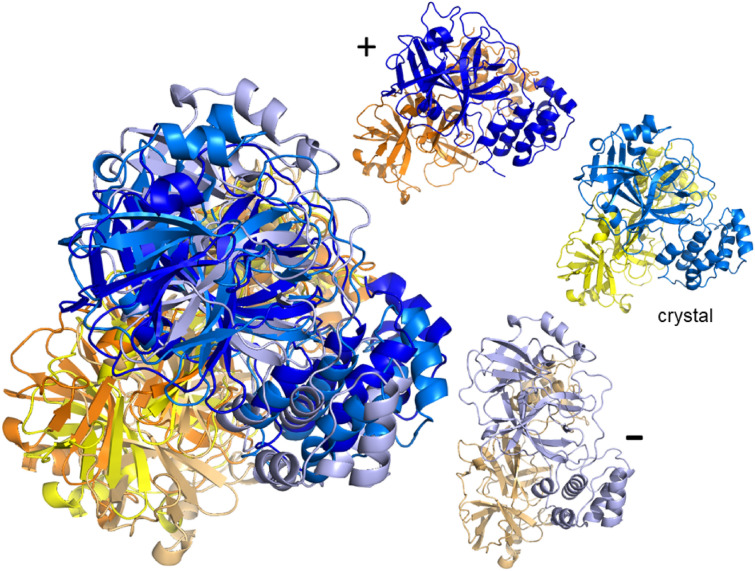
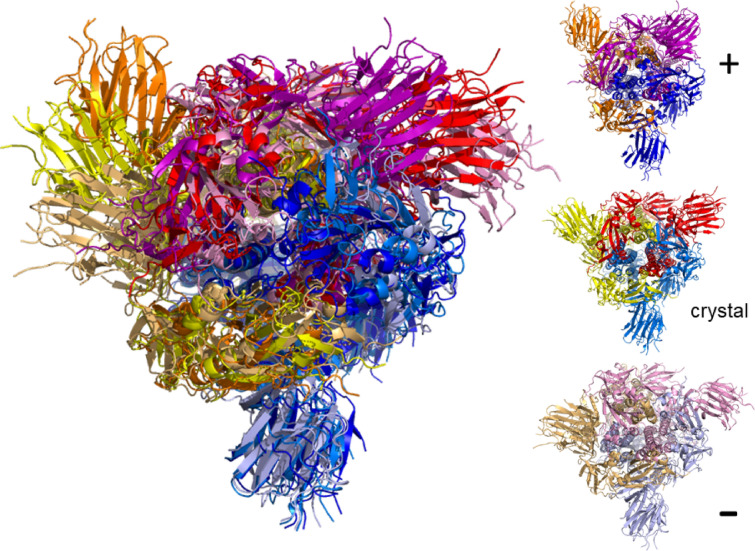
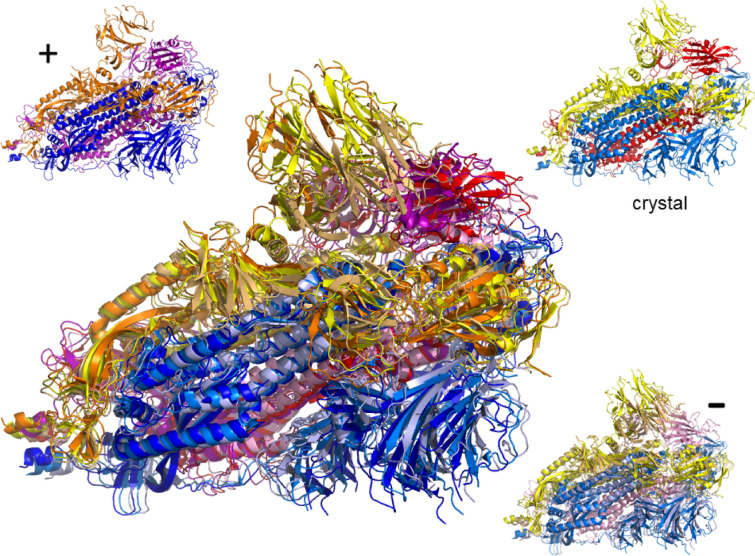
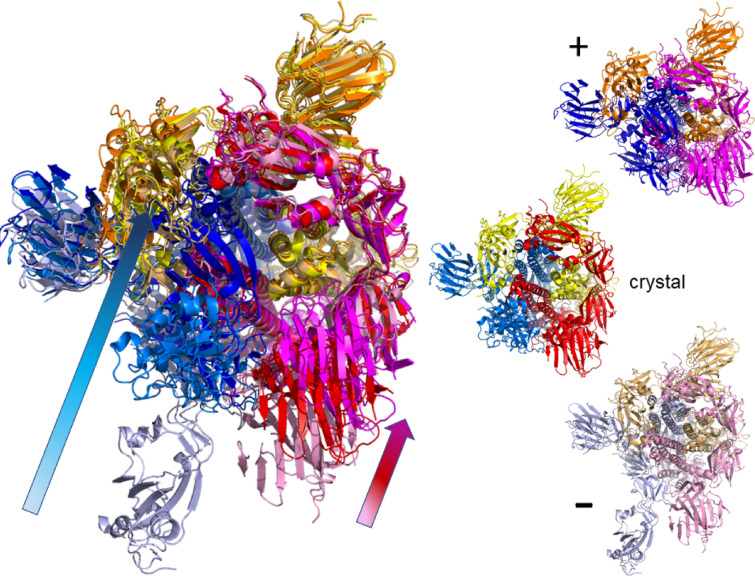
The worldwide CoVid-19 pandemic has led to an unprecedented push across the whole of the scientific community to develop a potent antiviral drug and vaccine as soon as possible. Existing academic, governmental and industrial institutions and companies have engaged in large-scale screening of existing drugs, in vitro, in vivo and in silico. Here, we are using in silico modelling of possible SARS-CoV-2 drug targets, as deposited on the Protein Databank (PDB), and ascertain their dynamics, flexibility and rigidity. For example, for the SARS-CoV-2 spike protein-using its complete homo-trimer configuration with 2905 residues-our method identifies a large-scale opening and closing of the S1 subunit through movement of the S[Formula: see text] domain. We compute the full structural information of this process, allowing for docking studies with possible drug structures. In a dedicated database, we present similarly detailed results for the further, nearly 300, thus far resolved SARS-CoV-2-related protein structures in the PDB.
全球范围内的 COVID-19 大流行促使整个科学界前所未有地加紧研发有效的抗病毒药物和疫苗。现有的学术、政府和工业机构及公司已大规模开展了对现有药物的筛选,包括体外、体内和计算机模拟筛选。在这里,我们利用已在蛋白质数据库(PDB)中注册的可能的 SARS-CoV-2 药物靶点的计算机模拟,确定它们的动态、灵活性和刚性。例如,对于 SARS-CoV-2 刺突蛋白,我们使用其带有 2905 个残基的完整同源三聚体结构,我们的方法通过 S[Formula: see text]结构域的运动来识别 S1 亚基的大规模开合。我们计算了这个过程的完整结构信息,允许与可能的药物结构进行对接研究。在一个专门的数据库中,我们为 PDB 中进一步的、迄今已解析的近 300 个 SARS-CoV-2 相关蛋白结构呈现了类似详细的结果。