Department of Medicine, EPPIcenter, University of California, San Francisco, CA, USA.
Chan Zuckerberg Biohub, San Francisco, CA, USA.
Malar J. 2021 Feb 26;20(1):116. doi: 10.1186/s12936-021-03630-4.
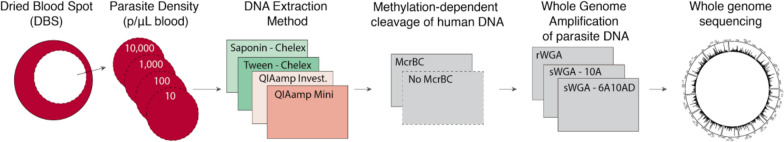
Whole-genome sequencing (WGS) is becoming increasingly useful to study the biology, epidemiology, and ecology of malaria parasites. Despite ease of sampling, DNA extracted from dried blood spots (DBS) has a high ratio of human DNA compared to parasite DNA, which poses a challenge for downstream genetic analyses. The effects of multiple methods for DNA extraction, digestion of methylated DNA, and amplification were evaluated on the quality and fidelity of WGS data recovered from DBS.
Low parasite density mock DBS samples were created, extracted either with Tween-Chelex or QIAamp, treated with or without McrBC, and amplified with one of three different amplification techniques (two sWGA primer sets and one rWGA). Extraction conditions were evaluated on performance of sequencing depth, percentiles of coverage, and expected SNP concordance.
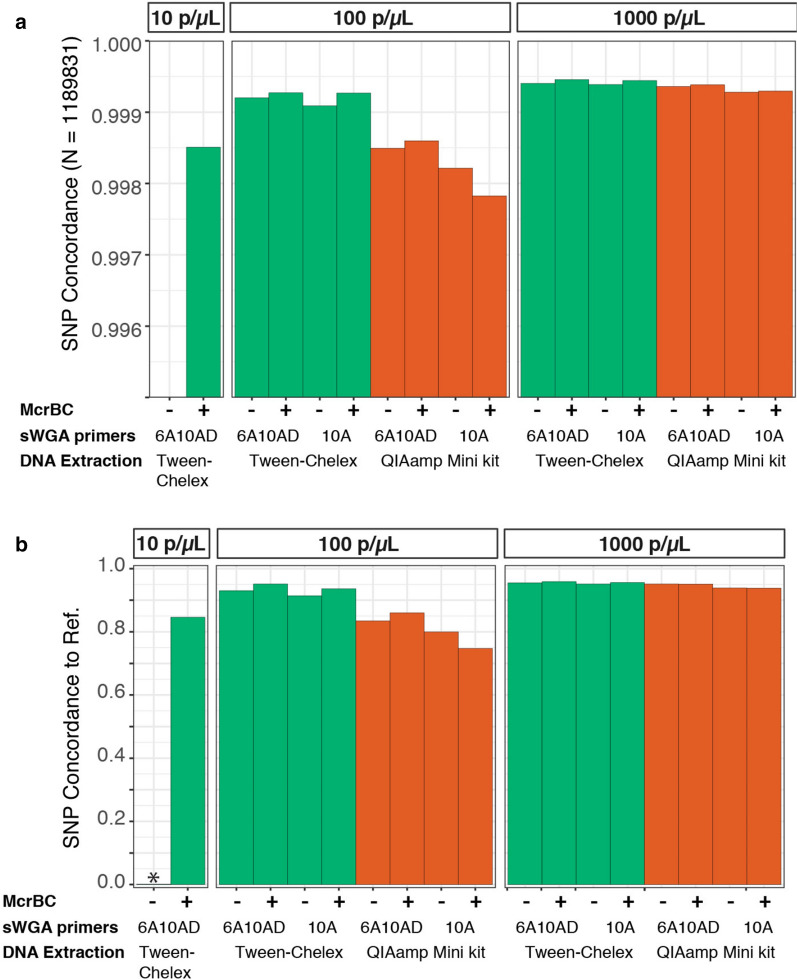
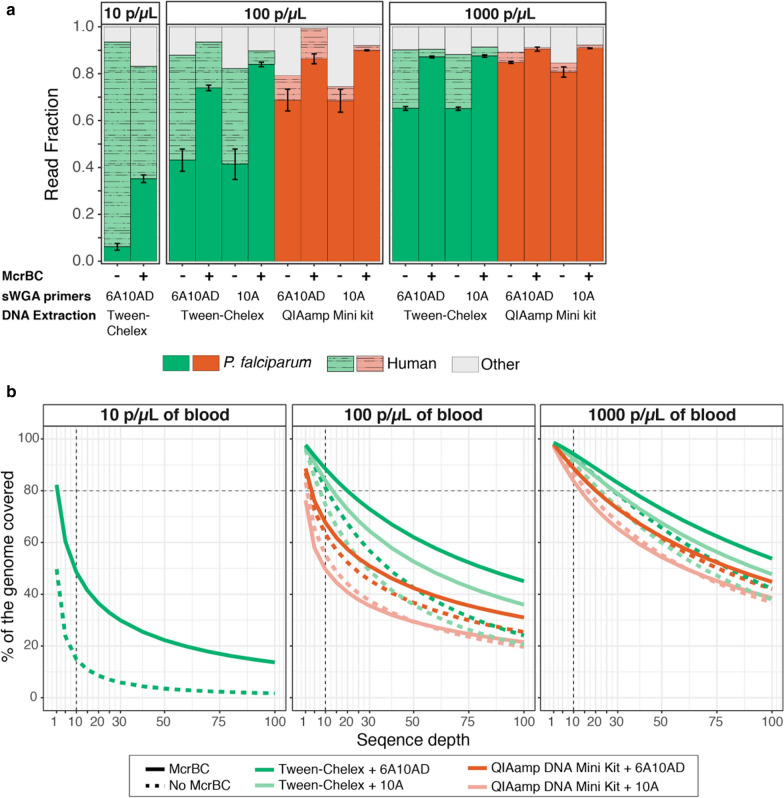
At 100 parasites/μL, Chelex-Tween-McrBC samples had higher coverage (5 × depth = 93% genome) than QIAamp extracted samples (5 × depth = 76% genome). The two evaluated sWGA primer sets showed minor differences in overall genome coverage and SNP concordance, with a newly proposed combination of 20 primers showing a modest improvement in coverage over those previously published.
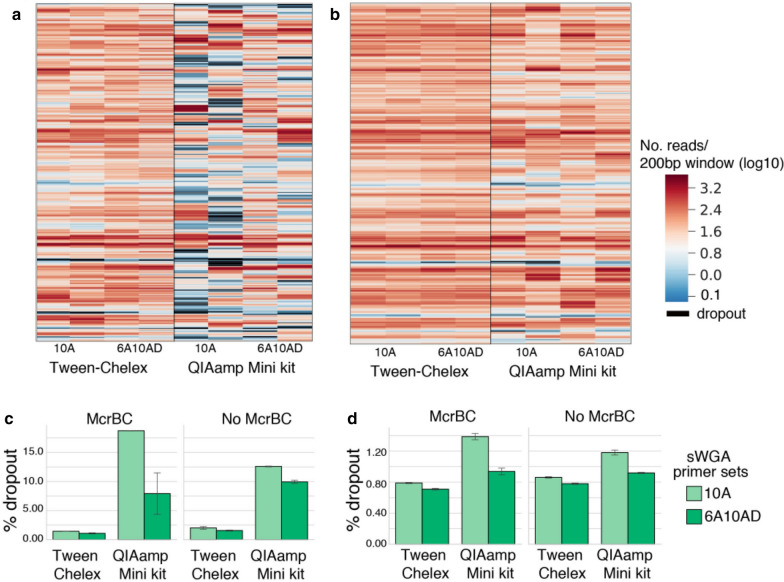
Overall, Tween-Chelex extracted samples that were treated with McrBC digestion and are amplified using 6A10AD sWGA conditions had minimal dropout rate, higher percentages of coverage at higher depth, and more accurate SNP concordance than QiaAMP extracted samples. These findings extend the results of previously reported methods, making whole genome sequencing accessible to a larger number of low density samples that are commonly encountered in cross-sectional surveys.
全基因组测序(WGS)在研究疟原虫的生物学、流行病学和生态学方面变得越来越有用。尽管采样方便,但与寄生虫 DNA 相比,从干血斑(DBS)中提取的 DNA 中人体 DNA 的比例很高,这给下游遗传分析带来了挑战。本研究评估了多种 DNA 提取方法、甲基化 DNA 消化以及扩增方法对从 DBS 中恢复的 WGS 数据的质量和保真度的影响。
创建低寄生虫密度模拟 DBS 样本,分别使用 Tween-Chelex 或 QIAamp 提取,用或不用 McrBC 处理,并使用三种不同的扩增技术(两种 sWGA 引物组和一种 rWGA)进行扩增。评估提取条件对测序深度、覆盖百分位数和预期 SNP 一致性的影响。
在 100 个寄生虫/μL 时,Chelex-Tween-McrBC 样本的覆盖度(5×深度=93%基因组)高于 QIAamp 提取样本(5×深度=76%基因组)。两种评估的 sWGA 引物组在总体基因组覆盖度和 SNP 一致性方面差异较小,一组 20 个新的引物组合在覆盖度上略有提高。
总体而言,用 McrBC 消化处理并用 6A10AD sWGA 条件扩增的 Tween-Chelex 提取样本具有最小的缺失率、更高的深度覆盖百分比和更准确的 SNP 一致性,优于 QiaAMP 提取样本。这些发现扩展了先前报道方法的结果,使全基因组测序更容易用于在横断面研究中常见的大量低密度样本。