Oyola Samuel O, Ariani Cristina V, Hamilton William L, Kekre Mihir, Amenga-Etego Lucas N, Ghansah Anita, Rutledge Gavin G, Redmond Seth, Manske Magnus, Jyothi Dushyanth, Jacob Chris G, Otto Thomas D, Rockett Kirk, Newbold Chris I, Berriman Matthew, Kwiatkowski Dominic P
Wellcome Trust Sanger Institute, Hinxton, CB10 1SA, UK.
International Livestock Research Institute, Box 30709, Nairobi, Kenya.
Malar J. 2016 Dec 20;15(1):597. doi: 10.1186/s12936-016-1641-7.
Translating genomic technologies into healthcare applications for the malaria parasite Plasmodium falciparum has been limited by the technical and logistical difficulties of obtaining high quality clinical samples from the field. Sampling by dried blood spot (DBS) finger-pricks can be performed safely and efficiently with minimal resource and storage requirements compared with venous blood (VB). Here, the use of selective whole genome amplification (sWGA) to sequence the P. falciparum genome from clinical DBS samples was evaluated, and the results compared with current methods that use leucodepleted VB.
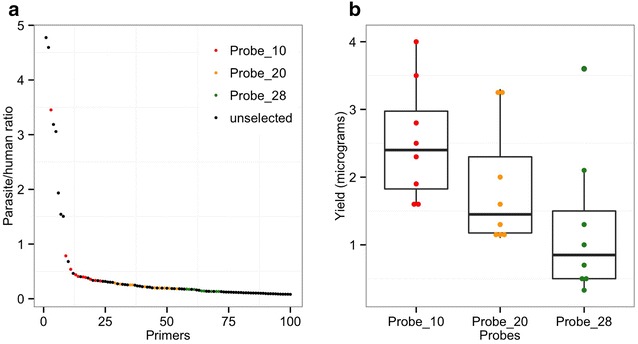
Parasite DNA with high (>95%) human DNA contamination was selectively amplified by Phi29 polymerase using short oligonucleotide probes of 8-12 mers as primers. These primers were selected on the basis of their differential frequency of binding the desired (P. falciparum DNA) and contaminating (human) genomes.
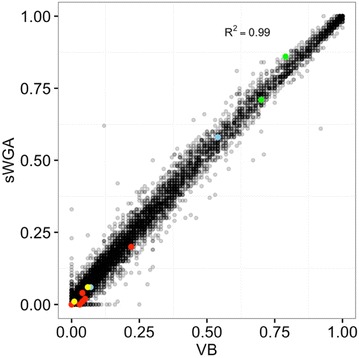
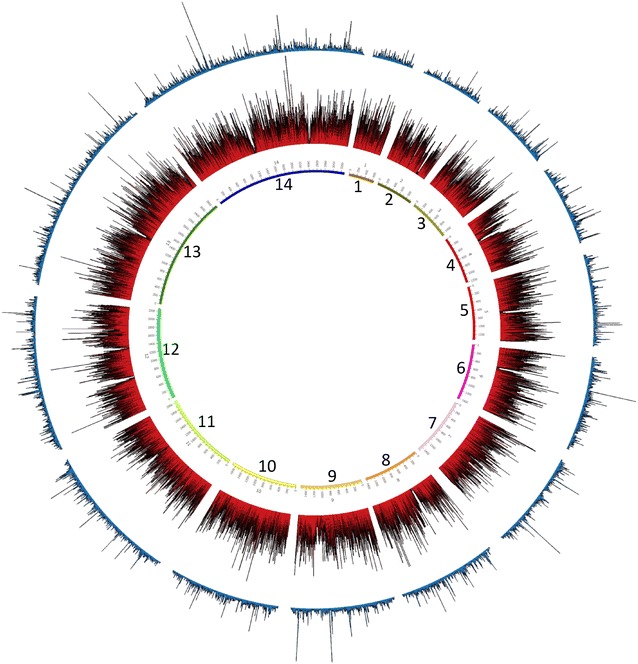
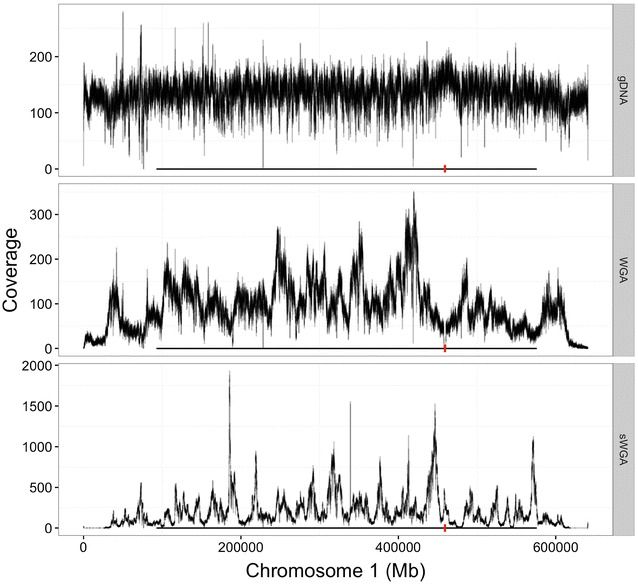
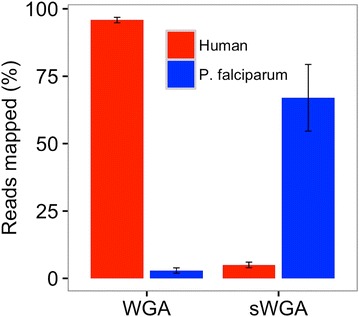
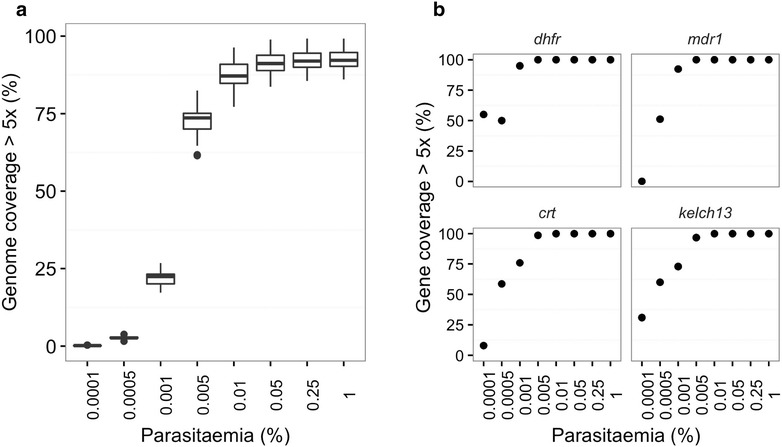
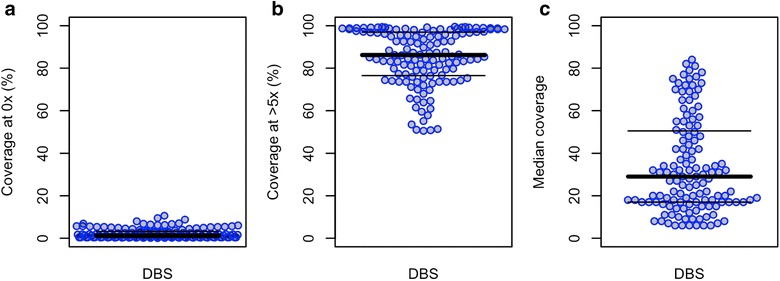
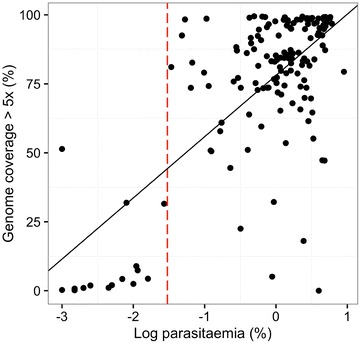
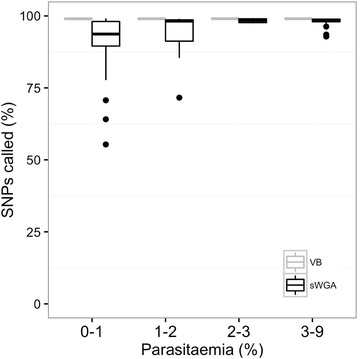
Using sWGA method, clinical samples from 156 malaria patients, including 120 paired samples for head-to-head comparison of DBS and leucodepleted VB were sequenced. Greater than 18-fold enrichment of P. falciparum DNA was achieved from DBS extracts. The parasitaemia threshold to achieve >5× coverage for 50% of the genome was 0.03% (40 parasites per 200 white blood cells). Over 99% SNP concordance between VB and DBS samples was achieved after excluding missing calls.
The sWGA methods described here provide a reliable and scalable way of generating P. falciparum genome sequence data from DBS samples. The current data indicate that it will be possible to get good quality sequence on most if not all drug resistance loci from the majority of symptomatic malaria patients. This technique overcomes a major limiting factor in P. falciparum genome sequencing from field samples, and paves the way for large-scale epidemiological applications.
将基因组技术转化为用于恶性疟原虫的医疗保健应用受到从现场获取高质量临床样本的技术和后勤困难的限制。与静脉血(VB)相比,通过干血斑(DBS)手指采血进行采样可以在资源和存储需求最小的情况下安全、高效地进行。在此,评估了使用选择性全基因组扩增(sWGA)对临床DBS样本中的恶性疟原虫基因组进行测序,并将结果与使用去白细胞VB的当前方法进行比较。
使用8-12聚体的短寡核苷酸探针作为引物,通过Phi29聚合酶选择性扩增人DNA污染率高(>95%)的寄生虫DNA。这些引物是根据它们与所需(恶性疟原虫DNA)和污染(人类)基因组结合的差异频率选择的。
使用sWGA方法,对156名疟疾患者的临床样本进行了测序,其中包括120对用于DBS和去白细胞VB头对头比较的配对样本。从DBS提取物中实现了恶性疟原虫DNA超过18倍的富集。实现基因组50%>5倍覆盖的疟原虫血症阈值为0.03%(每200个白细胞中有40个寄生虫)。排除缺失调用后,VB和DBS样本之间的单核苷酸多态性(SNP)一致性超过99%。
本文所述的sWGA方法提供了一种从DBS样本生成恶性疟原虫基因组序列数据的可靠且可扩展的方法。当前数据表明,从大多数有症状的疟疾患者中,即使不是全部耐药位点,也有可能获得高质量序列。该技术克服了从现场样本进行恶性疟原虫基因组测序的一个主要限制因素,并为大规模流行病学应用铺平了道路。