Malaria Research Program, Center for Vaccine Development and Global Health, University of Maryland School of Medicine, Baltimore, MD, USA.
Institute for Genome Sciences, University of Maryland School of Medicine, Baltimore, MD, USA.
Malar J. 2020 Mar 30;19(1):135. doi: 10.1186/s12936-020-03195-8.
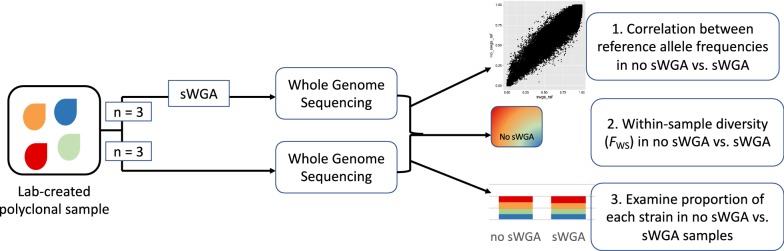
Owing to the large amount of host DNA in clinical samples, generation of high-quality Plasmodium falciparum whole genome sequencing (WGS) data requires enrichment for parasite DNA. Enrichment is often achieved by leukocyte depletion of infected blood prior to storage. However, leukocyte depletion is difficult in low-resource settings and limits analysis to prospectively-collected samples. As a result, approaches such as selective whole genome amplification (sWGA) are being used to enrich for parasite DNA. However, sWGA has had limited success in generating reliable sequencing data from low parasitaemia samples. In this study, enzymatic digestion with MspJI prior to sWGA and whole genome sequencing was evaluated to determine whether this approach improved genome coverage compared to sWGA alone. The potential of sWGA to cause amplification bias in polyclonal infections was also examined.
DNA extracted from laboratory-created dried blood spots was treated with a modification-dependent restriction endonuclease, MspJI, and filtered via vacuum filtration. Samples were then selectively amplified using a previously reported sWGA protocol and subjected to WGS. Genome coverage statistics were compared between the optimized sWGA approach and the previously reported sWGA approach performed in parallel. Differential amplification by sWGA was assessed by comparing WGS data generated from lab-created mixtures of parasite isolates, from the same geographical region, generated with or without sWGA.
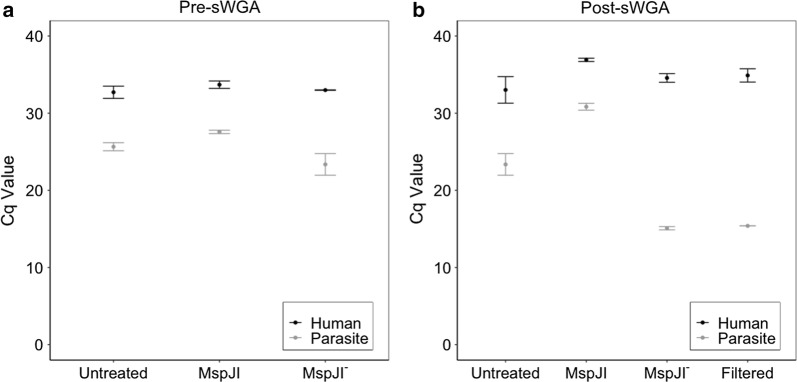
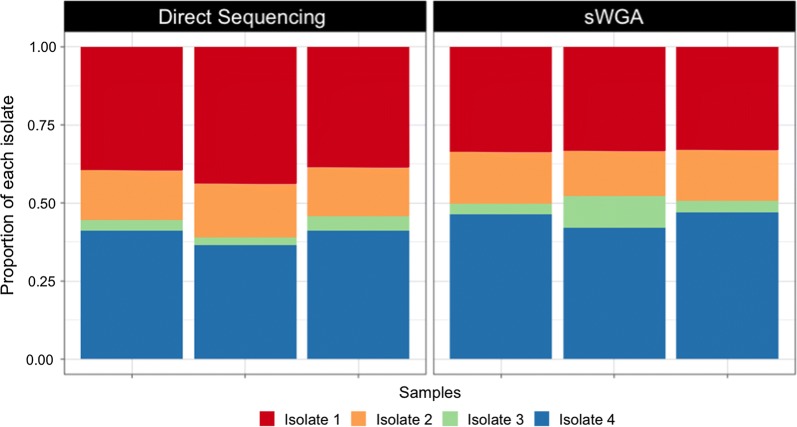
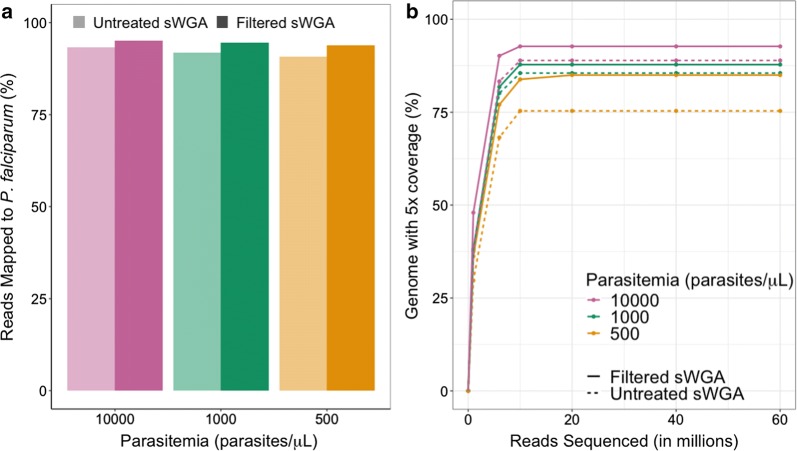
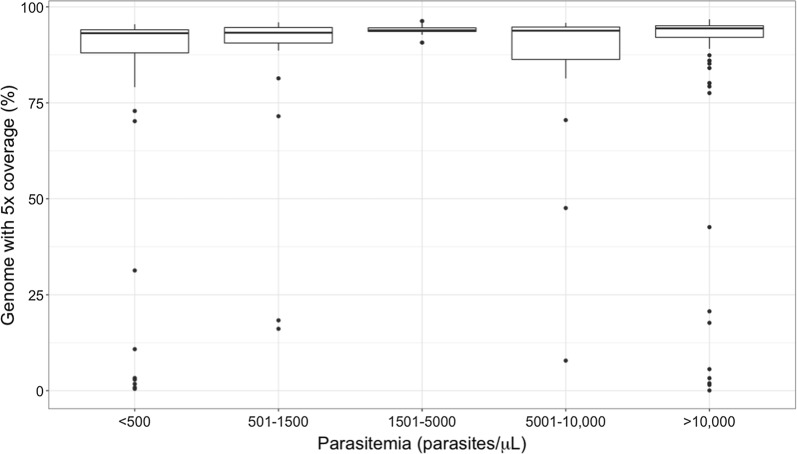
MspJI digestion did not enrich for parasite DNA. Samples that underwent vacuum filtration (without MspJI digestion) prior to sWGA had the highest parasite DNA concentration and displayed greater genome coverage compared to MspJI + sWGA and sWGA alone, particularly for low parasitaemia samples. The optimized sWGA (filtration + sWGA) approach was successfully used to generate WGS data from 218 non-leukocyte depleted field samples from Malawi. Sequences from lab-created mixtures of parasites did not show evidence of differential amplification of parasite strains compared to directly sequenced samples.
This optimized sWGA approach is a reliable method to obtain WGS data from non-leukocyte depleted, low parasitaemia samples. The absence of amplification bias in data generated from mixtures of isolates from the same geographic region suggests that this approach can be appropriately used for molecular epidemiological studies.
由于临床样本中存在大量宿主 DNA,因此需要对疟原虫全基因组测序 (WGS) 数据进行寄生虫 DNA 富集,才能生成高质量的数据。通常,在储存之前,通过白细胞去除来富集寄生虫 DNA。然而,在资源匮乏的环境中,白细胞去除较为困难,且会限制分析仅限于前瞻性采集的样本。因此,人们正在采用选择性全基因组扩增 (sWGA) 等方法来富集寄生虫 DNA。然而,sWGA 在从低寄生虫血症样本中生成可靠测序数据方面的效果有限。在这项研究中,我们评估了在 sWGA 和 WGS 之前用 MspJI 进行酶消化,以确定与单独进行 sWGA 相比,这种方法是否能提高基因组覆盖度。我们还研究了 sWGA 在多克隆感染中引起扩增偏倚的潜力。
从实验室制备的干血斑中提取的 DNA 用依赖修饰的限制内切酶 MspJI 处理,然后通过真空过滤。然后,使用之前报道的 sWGA 方案对样品进行选择性扩增,并进行 WGS。将优化后的 sWGA 方法与同时进行的之前报道的 sWGA 方法进行比较,以比较基因组覆盖度统计数据。通过比较使用或不使用 sWGA 生成的来自同一地理区域的寄生虫分离株的实验室创建混合物的 WGS 数据,评估 sWGA 的差异扩增。
MspJI 消化并未富集寄生虫 DNA。与 MspJI+sWGA 和 sWGA 单独处理相比,在进行 sWGA 之前进行真空过滤(不进行 MspJI 消化)的样品寄生虫 DNA 浓度最高,基因组覆盖度更高,尤其是对于低寄生虫血症样本。该优化的 sWGA(过滤+sWGA)方法成功地从马拉维的 218 个非白细胞去除的现场样本中生成了 WGS 数据。与直接测序样本相比,来自实验室创建的寄生虫混合物的序列没有显示出寄生虫株差异扩增的证据。
这种优化的 sWGA 方法是一种从非白细胞去除、低寄生虫血症样本中获取 WGS 数据的可靠方法。来自同一地理区域的分离株混合物生成的数据中不存在扩增偏倚,这表明该方法可以适当用于分子流行病学研究。