Elimu Informatics, 1160 Brickyard Cove Rd Ste 200, Richmond, CA, 94801-4173, USA.
Department of Pathology, Immunology and Laboratory Medicine, University of Florida, Gainesville, FL, USA.
BMC Bioinformatics. 2021 Mar 2;22(1):104. doi: 10.1186/s12859-021-04039-1.
VCF formatted files are the lingua franca of next-generation sequencing, whereas HL7 FHIR is emerging as a standard language for electronic health record interoperability. A growing number of FHIR-based clinical genomics applications are emerging. Here, we describe an open source utility for converting variants from VCF format into HL7 FHIR format.
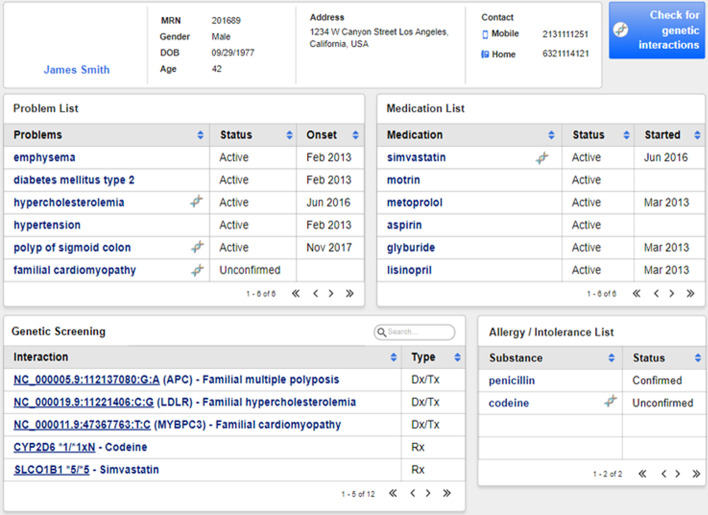
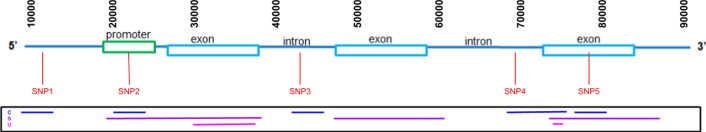
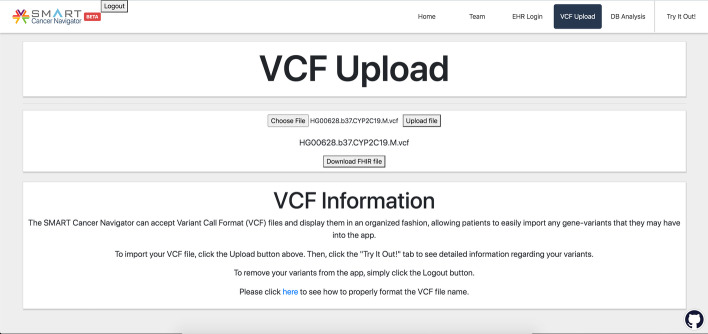
vcf2fhir converts VCF variants into a FHIR Genomics Diagnostic Report. Conversion translates each VCF row into a corresponding FHIR-formatted variant in the generated report. In scope are simple variants (SNVs, MNVs, Indels), along with zygosity and phase relationships, for autosomes, sex chromosomes, and mitochondrial DNA. Input parameters include VCF file and genome build ('GRCh37' or 'GRCh38'); and optionally a conversion region that indicates the region(s) to convert, a studied region that lists genomic regions studied by the lab, and a non-callable region that lists studied regions deemed uncallable by the lab. Conversion can be limited to a subset of VCF by supplying genomic coordinates of the conversion region(s). If studied and non-callable regions are also supplied, the output FHIR report will include 'region-studied' observations that detail which portions of the conversion region were studied, and of those studied regions, which portions were deemed uncallable. We illustrate the vcf2fhir utility via two case studies. The first, 'SMART Cancer Navigator', is a web application that offers clinical decision support by linking patient EHR information to cancerous gene variants. The second, 'Precision Genomics Integration Platform', intersects a patient's FHIR-formatted clinical and genomic data with knowledge bases in order to provide on-demand delivery of contextually relevant genomic findings and recommendations to the EHR.
Experience to date shows that the vcf2fhir utility can be effectively woven into clinically useful genomic-EHR integration pipelines. Additional testing will be a critical step towards the clinical validation of this utility, enabling it to be integrated in a variety of real world data flow scenarios. For now, we propose the use of this utility primarily to accelerate FHIR Genomics understanding and to facilitate experimentation with further integration of genomics data into the EHR.
VCF 格式文件是下一代测序的通用语言,而 HL7 FHIR 正逐渐成为电子健康记录互操作性的标准语言。越来越多基于 FHIR 的临床基因组学应用正在出现。在这里,我们描述了一种将变体从 VCF 格式转换为 HL7 FHIR 格式的开源实用程序。
vcf2fhir 将 VCF 变体转换为 FHIR 基因组诊断报告。转换将每个 VCF 行转换为生成报告中相应的 FHIR 格式变体。范围包括常染色体、性染色体和线粒体 DNA 的简单变体(SNV、MNV、插入缺失),以及同型合子和相位关系。输入参数包括 VCF 文件和基因组构建('GRCh37'或'GRCh38');以及可选的转换区域,该区域指示要转换的区域,研究区域列出实验室研究的基因组区域,以及不可调用区域,该区域列出实验室认为不可调用的研究区域。通过提供转换区域的基因组坐标,可以将转换限制为 VCF 的子集。如果还提供了研究和不可调用的区域,则输出 FHIR 报告将包括“区域研究”观察结果,详细说明转换区域的哪些部分进行了研究,以及在研究区域中,哪些部分被认为不可调用。我们通过两个案例研究来说明 vcf2fhir 实用程序。第一个是“SMART Cancer Navigator”,这是一个提供临床决策支持的 Web 应用程序,通过将患者的 EHR 信息与癌症基因变体链接来实现。第二个是“Precision Genomics Integration Platform”,它将患者的 FHIR 格式的临床和基因组数据与知识库交叉,以便根据需要将上下文相关的基因组发现和建议提供给 EHR。
迄今为止的经验表明,vcf2fhir 实用程序可以有效地编织到临床有用的基因组-EHR 集成管道中。进一步的测试将是该实用程序临床验证的关键步骤,使其能够集成到各种实际数据流场景中。目前,我们建议主要使用此实用程序来加速对 FHIR 基因组的理解,并促进进一步将基因组数据集成到 EHR 中的实验。