Gertig Christoph, Erdkamp Eric, Ernst Andreas, Hemprich Carl, Kröger Leif C, Langanke Jens, Bardow André, Leonhard Kai
Institute of Technical Thermodynamics, RWTH Aachen University, Schinkelstraße 8, 52062, Aachen, Germany.
CAT Catalytic Center, RWTH Aachen University, Worringerweg 2, 52074, Aachen, Germany.
ChemistryOpen. 2021 May;10(5):534-544. doi: 10.1002/open.202000150. Epub 2021 Mar 3.
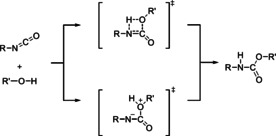
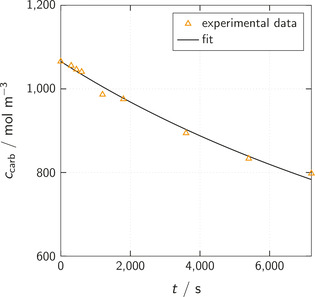
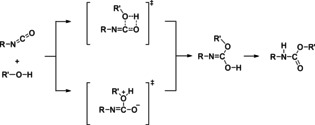
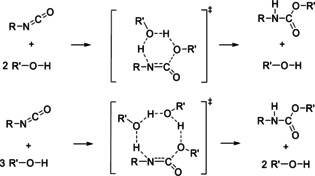
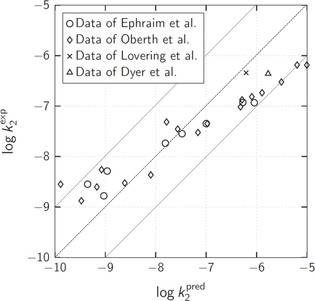
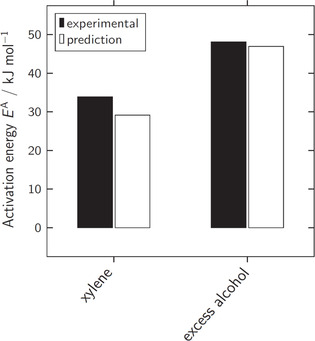
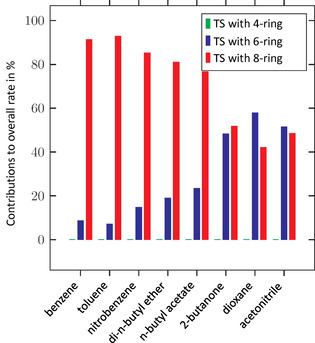
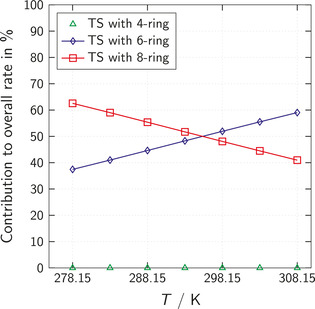
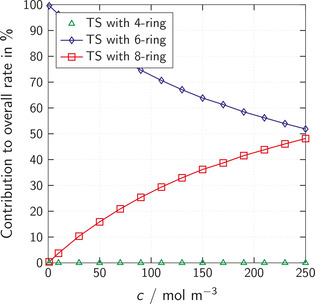
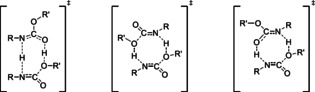
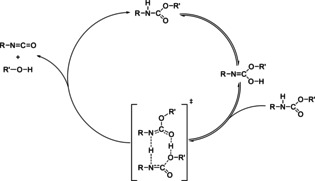
The chemistry of urethanes plays a key role in important industrial processes. Although catalysts are often used, the study of the reactions without added catalysts provides the basis for a deeper understanding. For the non-catalytic urethane formation and cleavage reactions, the dominating reaction mechanism has long been debated. To our knowledge, the reaction kinetics have not been predicted quantitatively so far. Therefore, we report a new computational study of urethane formation and cleavage reactions. To analyze various potential reaction mechanisms and to predict the reaction rate constants quantum chemistry and transition state theory were employed. For validation, experimental data from literature and from own experiments were used. Quantitative agreement of experiments and predictions could be demonstrated. The calculations confirm earlier assumptions that urethane formation reactions proceed via mechanisms where alcohol molecules act as auto-catalysts. Our results show that it is essential to consider several transition states corresponding to different reaction orders to enable agreement with experimental observations. Urethane cleavage seems to be catalyzed by an isourethane, leading to an observed 2nd-order dependence of the reaction rate on the urethane concentration. The results of our study support a deeper understanding of the reactions as well as a better description of reaction kinetics and will therefore help in catalyst development and process optimization.
聚氨酯的化学性质在重要的工业过程中起着关键作用。尽管经常使用催化剂,但对无添加催化剂时反应的研究为更深入的理解提供了基础。对于非催化聚氨酯的形成和裂解反应,主导反应机理长期以来一直存在争议。据我们所知,到目前为止,反应动力学尚未得到定量预测。因此,我们报告了一项关于聚氨酯形成和裂解反应的新计算研究。为了分析各种潜在的反应机理并预测反应速率常数,采用了量子化学和过渡态理论。为了进行验证,使用了文献和我们自己实验中的实验数据。实验和预测之间的定量一致性得到了证明。计算结果证实了早期的假设,即聚氨酯形成反应通过醇分子作为自催化剂的机理进行。我们的结果表明,必须考虑对应于不同反应级数的几个过渡态,才能与实验观察结果一致。聚氨酯裂解似乎由异氰酸酯催化,导致观察到反应速率对聚氨酯浓度的二级依赖性。我们的研究结果有助于更深入地理解这些反应,并更好地描述反应动力学,因此将有助于催化剂开发和工艺优化。