Edeleva Mariya, Van Steenberge Paul H M, Sabbe Maarten K, D'hooge Dagmar R
Laboratory for Chemical Technology (LCT), Ghent University, Technologiepark 125, 9052 Zwijnaarde, Belgium.
Industrial Catalysis and Adsorption Technology (INCAT), Ghent University, Valentin Vaerwyckweg 1, 9000 Ghent, Belgium.
Polymers (Basel). 2021 Sep 7;13(18):3027. doi: 10.3390/polym13183027.
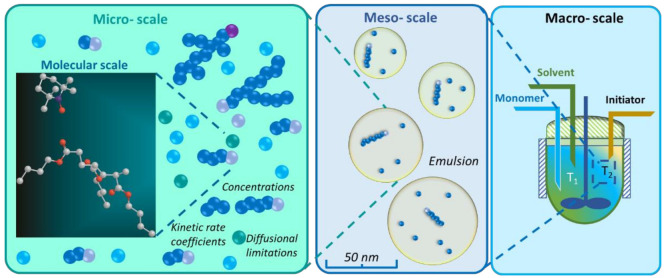
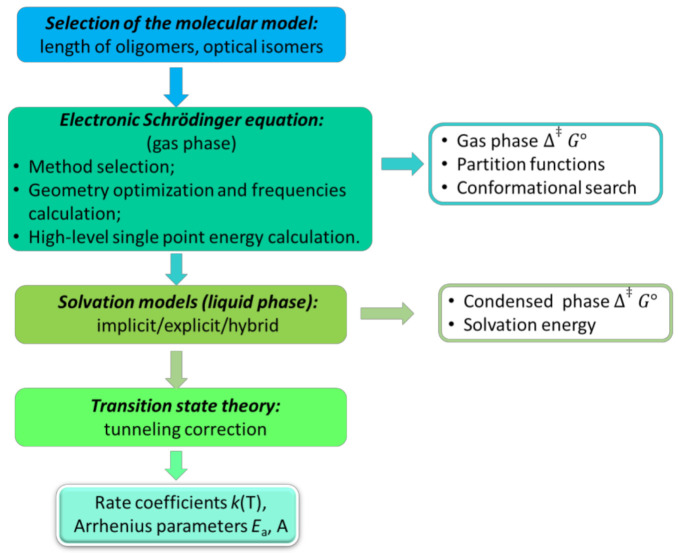
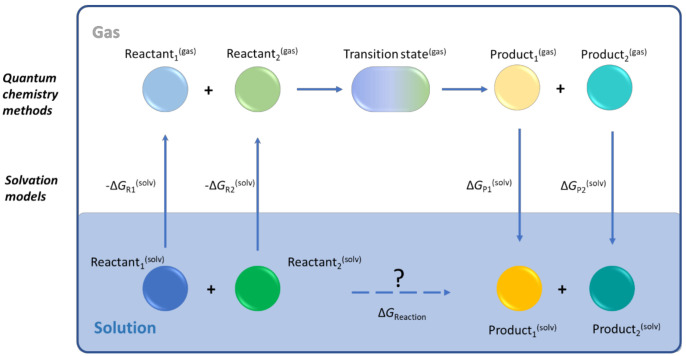
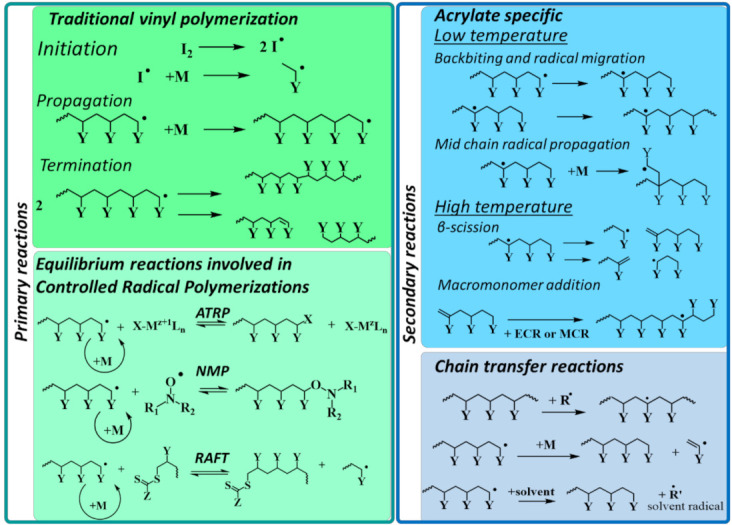
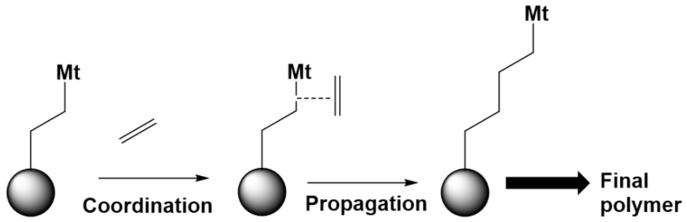
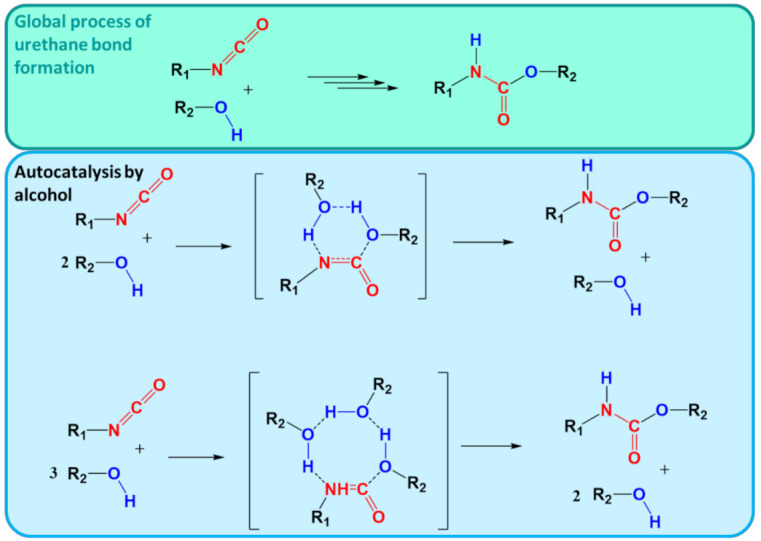
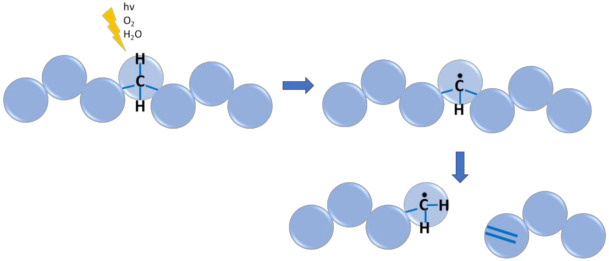
In recent decades, quantum chemical calculations (QCC) have increased in accuracy, not only providing the ranking of chemical reactivities and energy barriers (e.g., for optimal selectivities) but also delivering more reliable equilibrium and (intrinsic/chemical) rate coefficients. This increased reliability of kinetic parameters is relevant to support the predictive character of kinetic modeling studies that are addressing actual concentration changes during chemical processes, taking into account competitive reactions and mixing heterogeneities. In the present contribution, guidelines are formulated on how to bridge the fields of computational chemistry and chemical kinetics. It is explained how condensed phase systems can be described based on conventional gas phase computational chemistry calculations. Case studies are included on polymerization kinetics, considering free and controlled radical polymerization, ionic polymerization, and polymer degradation. It is also illustrated how QCC can be directly linked to material properties.
近几十年来,量子化学计算(QCC)的准确性有所提高,不仅能够提供化学反应活性和能垒的排序(例如用于优化选择性),还能给出更可靠的平衡和(本征/化学)速率系数。动力学参数可靠性的提高有助于支持动力学建模研究的预测性,这些研究考虑了竞争反应和混合不均匀性,旨在解决化学过程中实际浓度的变化。在本论文中,制定了关于如何弥合计算化学和化学动力学领域的指导方针。解释了如何基于传统的气相计算化学计算来描述凝聚相体系。包含了关于聚合动力学的案例研究,涉及自由基聚合、可控自由基聚合、离子聚合以及聚合物降解。还阐述了量子化学计算如何能够直接与材料性能相关联。