Grupo de Química Computacional y Teórica (QCT-USFQ), Departamento de Ingeniería Química, Colegio Politécnico, Universidad San Francisco de Quito, Diego de Robles y Vía Interoceánica, Quito 170901, Ecuador.
Grupo de Investigaciones en Química y Biología, Departamento de Química y Biología, Facultad de Ciencias Exactas, Universidad del Norte, Carrera 51B, Km 5, vía Puerto Colombia, Barranquilla 081007, Colombia.
Molecules. 2021 Feb 19;26(4):1100. doi: 10.3390/molecules26041100.
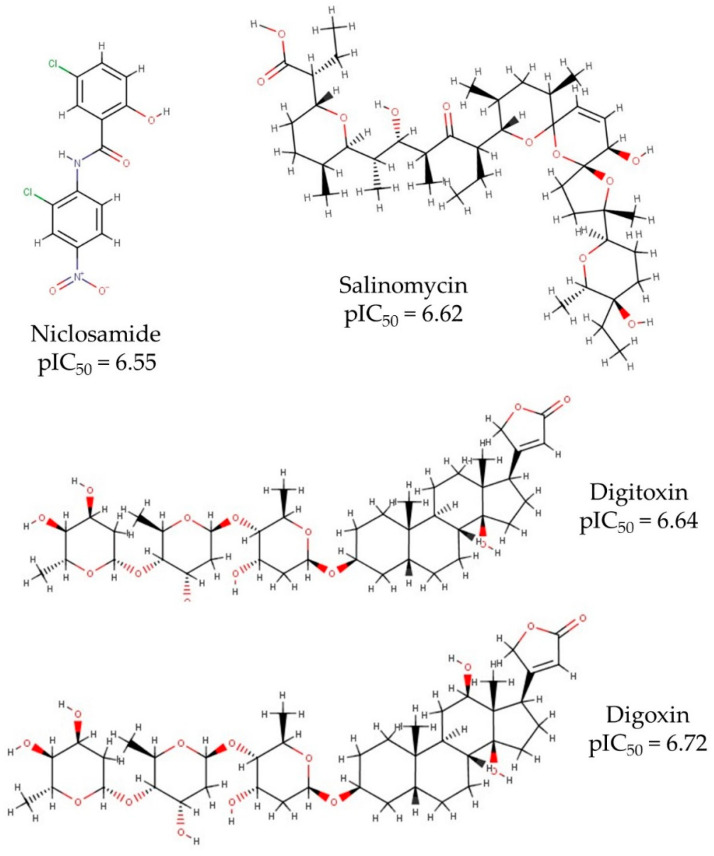
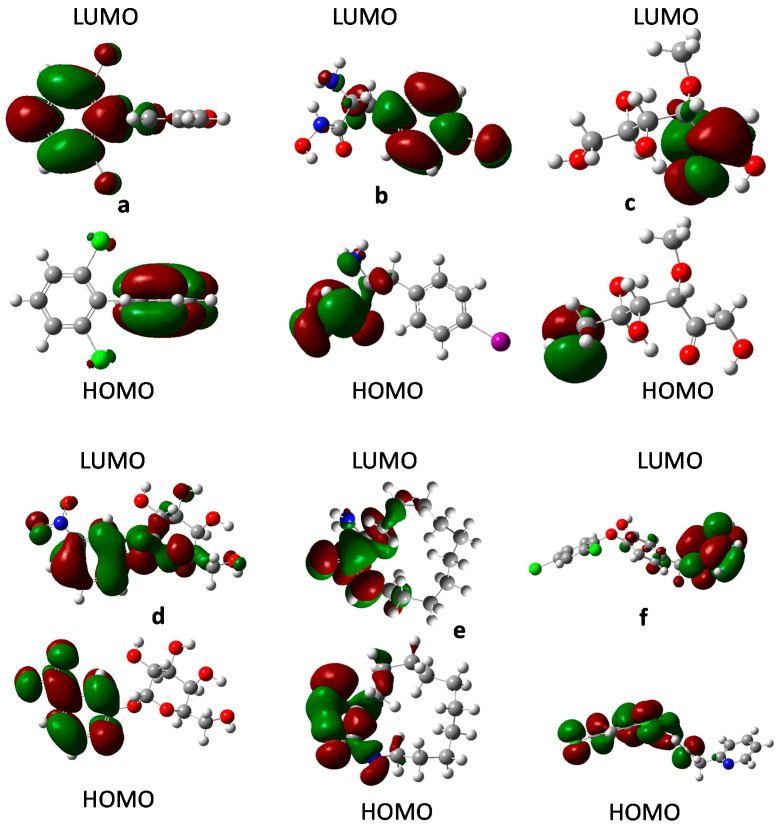
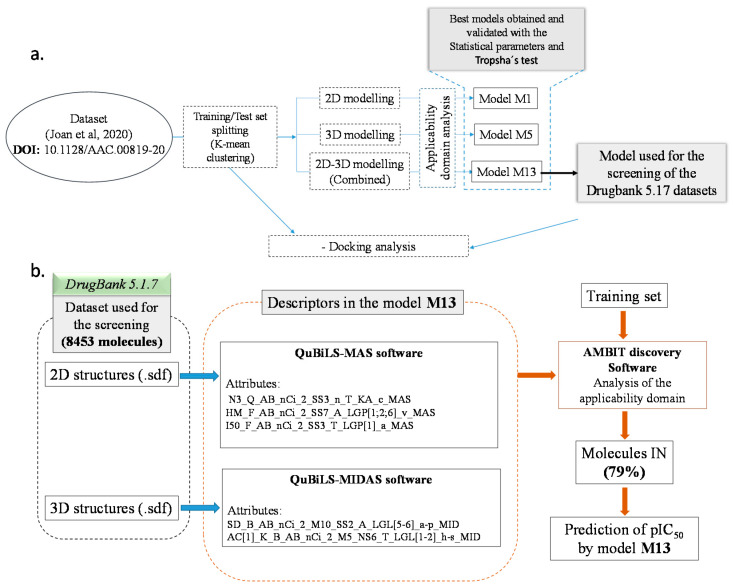
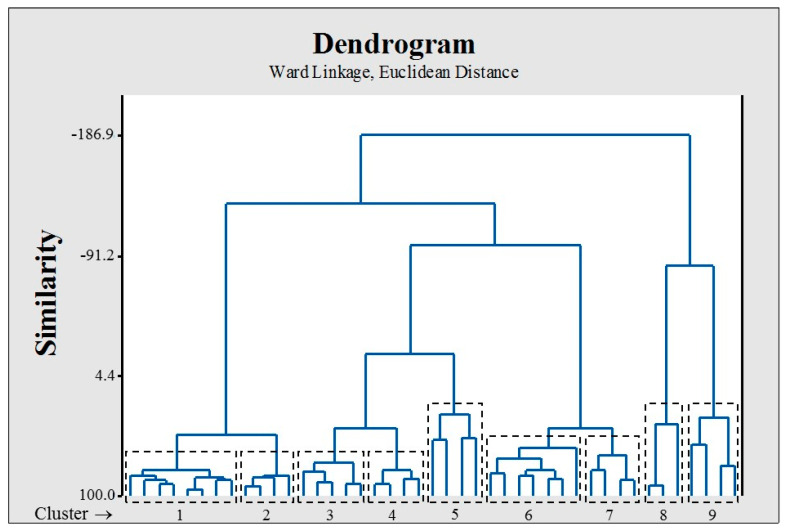
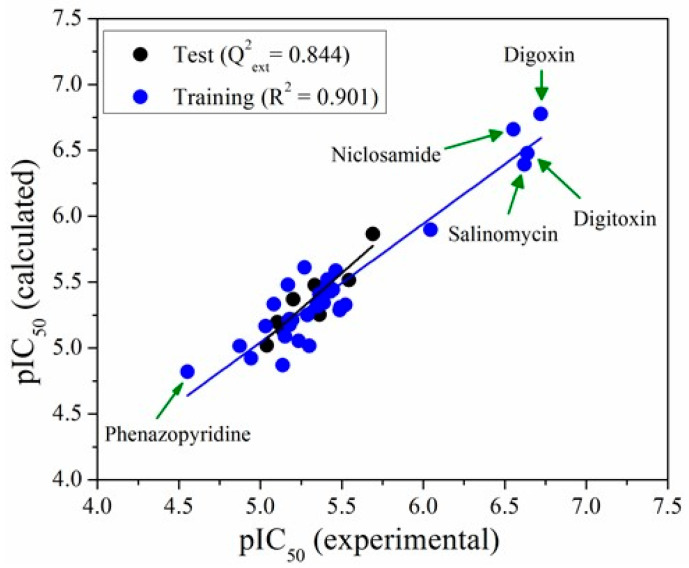
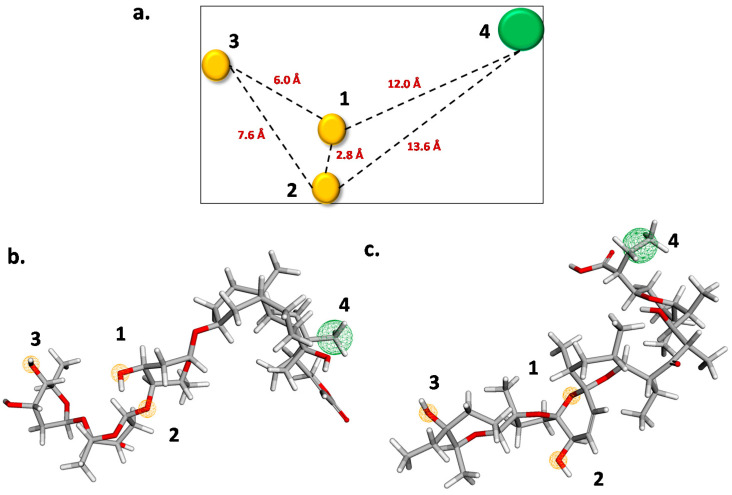
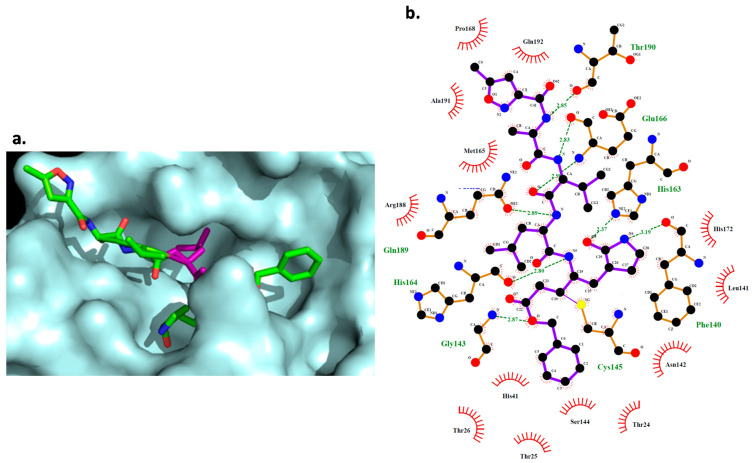
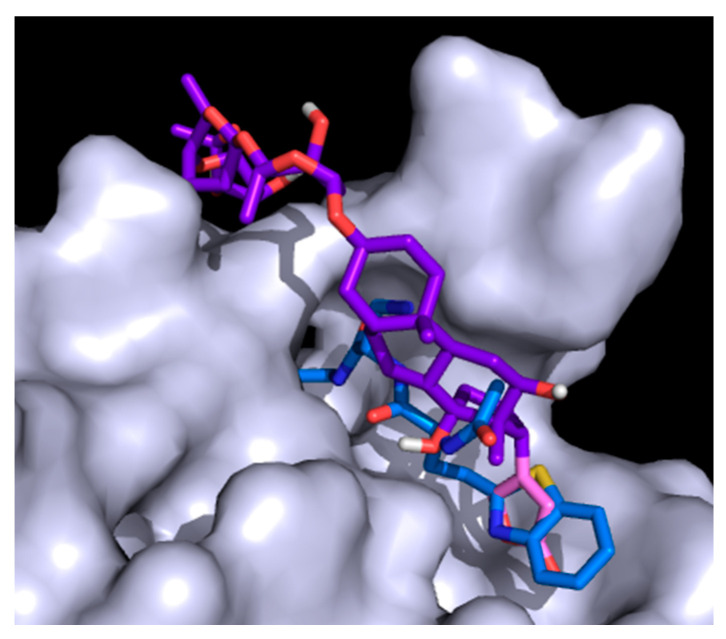
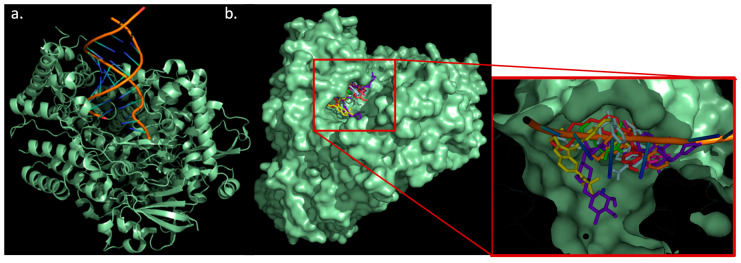
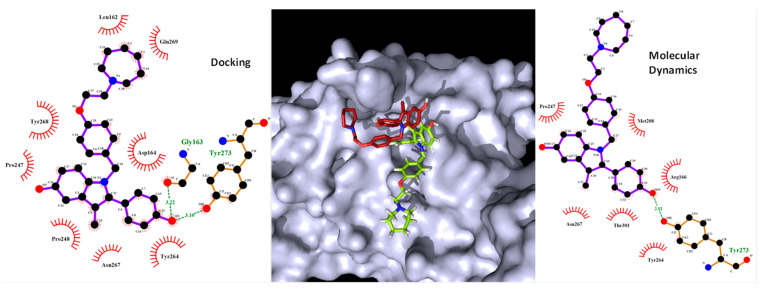
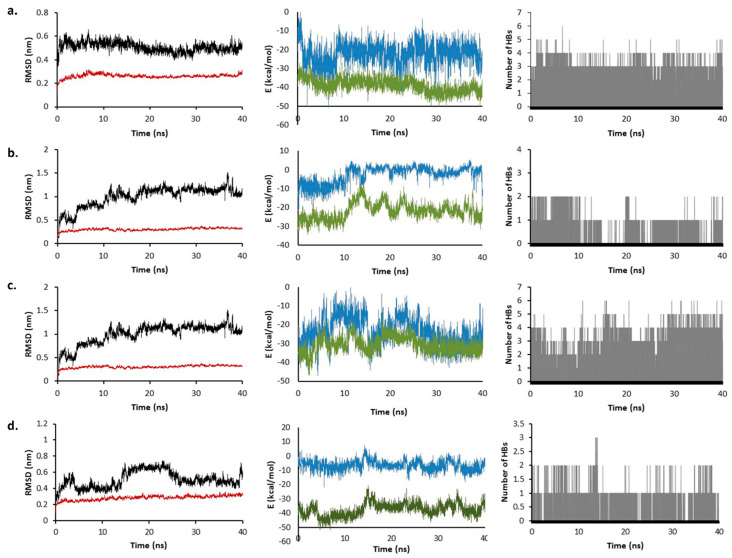
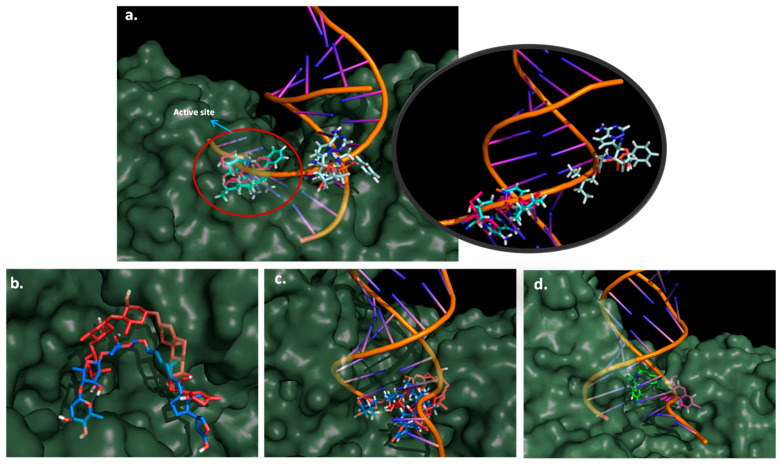
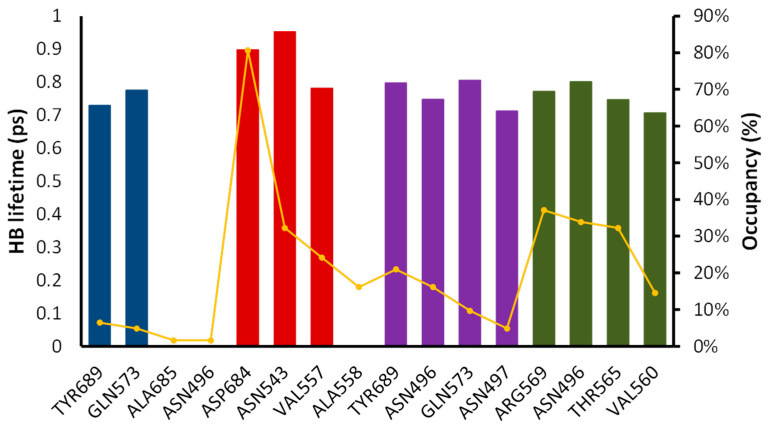
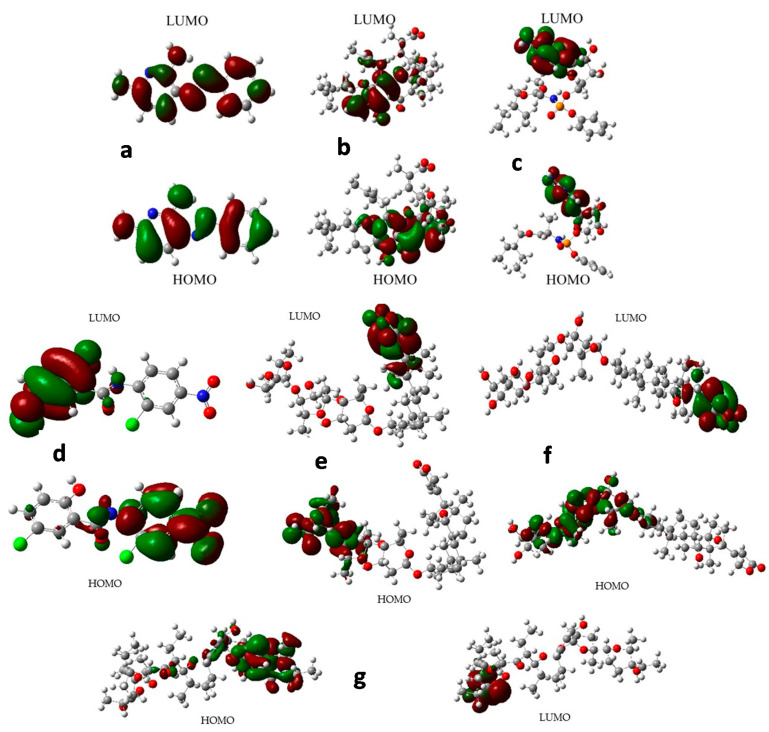
Coronavirus desease 2019 (COVID-19) is responsible for more than 1.80 M deaths worldwide. A Quantitative Structure-Activity Relationships (QSAR) model is developed based on experimental pIC values reported for a structurally diverse dataset. A robust model with only five descriptors is found, with values of R = 0.897, Q = 0.854, and Q = 0.876 and complying with all the parameters established in the validation Tropsha's test. The analysis of the applicability domain (AD) reveals coverage of about 90% for the external test set. Docking and molecular dynamic analysis are performed on the three most relevant biological targets for SARS-CoV-2: main protease, papain-like protease, and RNA-dependent RNA polymerase. A screening of the DrugBank database is executed, predicting the pIC value of 6664 drugs, which are IN the AD of the model (coverage = 79%). Fifty-seven possible potent anti-COVID-19 candidates with pIC values > 6.6 are identified, and based on a pharmacophore modelling analysis, four compounds of this set can be suggested as potent candidates to be potential inhibitors of SARS-CoV-2. Finally, the biological activity of the compounds was related to the frontier molecular orbitals shapes.
2019 年冠状病毒病(COVID-19)在全球范围内造成超过 180 万人死亡。基于结构多样的数据集报告的实验 pIC 值,开发了一种定量构效关系(QSAR)模型。发现了一个只有五个描述符的稳健模型,其 R 值为 0.897,Q 值为 0.854,Q 值为 0.876,并且符合 Tropsha 验证测试中建立的所有参数。适用性域(AD)的分析表明,外部测试集的覆盖率约为 90%。对 SARS-CoV-2 的三个最重要的生物学靶标:主要蛋白酶、木瓜蛋白酶样蛋白酶和 RNA 依赖性 RNA 聚合酶进行对接和分子动力学分析。对 DrugBank 数据库进行了筛选,预测了 6664 种药物的 pIC 值,这些药物都在模型的 AD 内(覆盖率=79%)。确定了 57 种可能具有 pIC 值>6.6 的有效抗 COVID-19 候选药物,并且基于药效团建模分析,可以提出该组中的四种化合物作为 SARS-CoV-2 的潜在抑制剂的有效候选药物。最后,将化合物的生物活性与前沿分子轨道形状相关联。