Department of Pharmaceutical Microbiology, Faculty of Pharmacy, Medical University of Gdańsk, 80-416 Gdańsk, Poland.
Department of Biology and Medical Genetics, Medical University of Gdańsk, 80-210 Gdańsk, Poland.
Int J Mol Sci. 2021 Feb 18;22(4):2033. doi: 10.3390/ijms22042033.
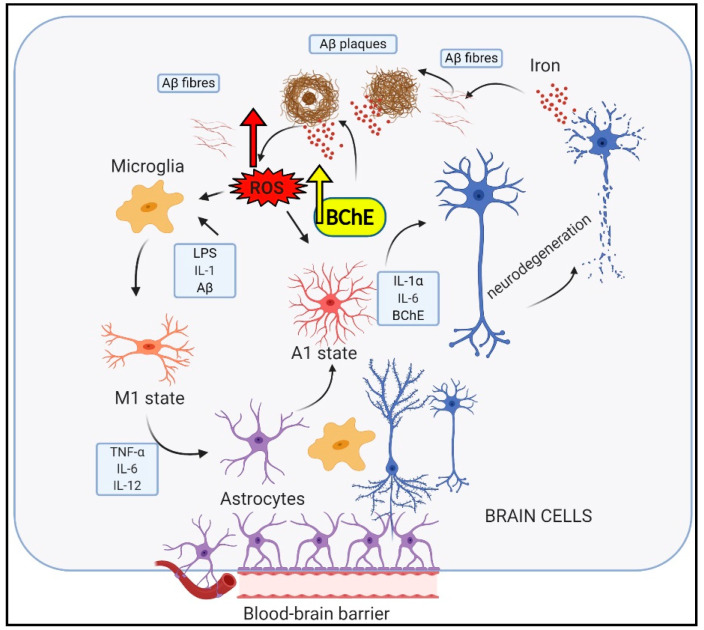
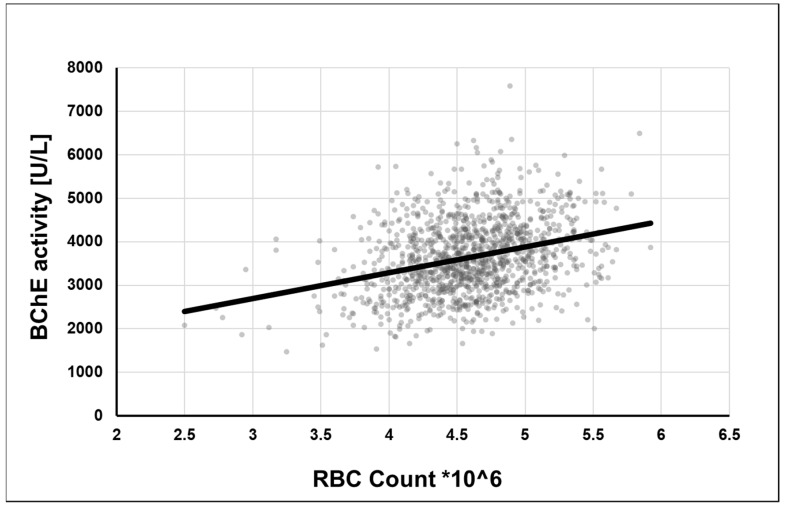
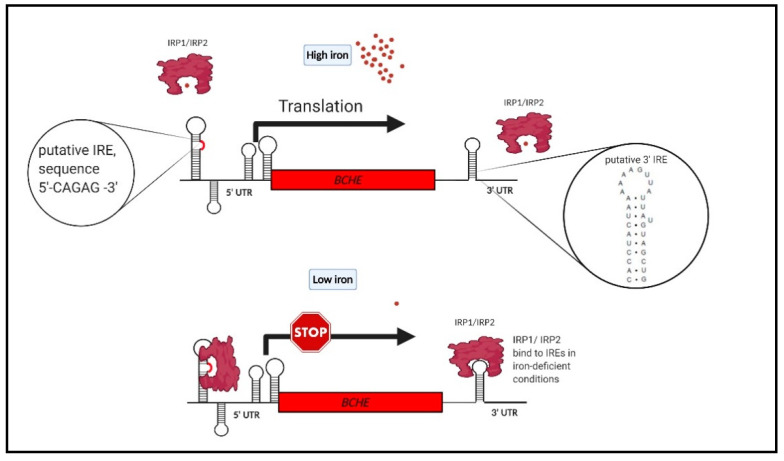
Alzheimer's disease (AD), the most common form of dementia in elderly individuals, is marked by progressive neuron loss. Despite more than 100 years of research on AD, there is still no treatment to cure or prevent the disease. High levels of amyloid-β (Aβ) plaques and neurofibrillary tangles (NFTs) in the brain are neuropathological hallmarks of AD. However, based on postmortem analyses, up to 44% of individuals have been shown to have high Aβ deposits with no clinical signs, due to having a "cognitive reserve". The biochemical mechanism explaining the prevention of cognitive impairment in the presence of Aβ plaques is still unknown. It seems that in addition to protein aggregation, neuroinflammatory changes associated with aging are present in AD brains that are correlated with a higher level of brain iron and oxidative stress. It has been shown that iron accumulates around amyloid plaques in AD mouse models and postmortem brain tissues of AD patients. Iron is required for essential brain functions, including oxidative metabolism, myelination, and neurotransmitter synthesis. However, an imbalance in brain iron homeostasis caused by aging underlies many neurodegenerative diseases. It has been proposed that high iron levels trigger an avalanche of events that push the progress of the disease, accelerating cognitive decline. Patients with increased amyloid plaques and iron are highly likely to develop dementia. Our observations indicate that the butyrylcholinesterase (BChE) level seems to be iron-dependent, and reports show that BChE produced by reactive astrocytes can make cognitive functions worse by accelerating the decay of acetylcholine in aging brains. Why, even when there is a genetic risk, do symptoms of the disease appear after many years? Here, we discuss the relationship between genetic factors, age-dependent iron tissue accumulation, and inflammation, focusing on AD.
阿尔茨海默病(AD)是老年人中最常见的痴呆症形式,其特征是神经元进行性丧失。尽管对 AD 进行了 100 多年的研究,但仍没有治愈或预防该疾病的方法。大脑中高水平的淀粉样蛋白-β(Aβ)斑块和神经原纤维缠结(NFTs)是 AD 的神经病理学标志。然而,根据死后分析,高达 44%的人表现出高 Aβ 沉积而没有临床症状,这是由于存在“认知储备”。解释存在 Aβ 斑块时预防认知障碍的生化机制仍不清楚。似乎除了蛋白聚集之外,与衰老相关的神经炎症变化也存在于 AD 大脑中,与大脑铁和氧化应激水平升高有关。已经表明,在 AD 小鼠模型和 AD 患者的死后脑组织中,铁会在淀粉样斑块周围积累。铁是包括氧化代谢、髓鞘形成和神经递质合成在内的大脑基本功能所必需的。然而,衰老导致的大脑铁稳态失衡是许多神经退行性疾病的基础。有人提出,高水平的铁会引发一系列事件,推动疾病的进展,加速认知能力下降。淀粉样斑块和铁含量增加的患者极有可能发展为痴呆症。我们的观察表明,丁酰胆碱酯酶(BChE)水平似乎依赖于铁,并且有报道表明,反应性星形胶质细胞产生的 BChE 可以通过加速衰老大脑中乙酰胆碱的降解使认知功能恶化。为什么即使存在遗传风险,疾病症状也要多年后才会出现?在这里,我们讨论了遗传因素、年龄相关的铁组织积累和炎症之间的关系,重点是 AD。