Liu-Wei Wang, Kafkas Şenay, Chen Jun, Dimonaco Nicholas J, Tegnér Jesper, Hoehndorf Robert
Computer, Electrical and Mathematical Sciences and Engineering Division, King Abdullah University of Science and Technology, Thuwal 23955, Saudi Arabia.
Computational Bioscience Research Center, King Abdullah University of Science and Technology, Thuwal 23955, Saudi Arabia.
Bioinformatics. 2021 Sep 9;37(17):2722-2729. doi: 10.1093/bioinformatics/btab147.
Infectious diseases caused by novel viruses have become a major public health concern. Rapid identification of virus-host interactions can reveal mechanistic insights into infectious diseases and shed light on potential treatments. Current computational prediction methods for novel viruses are based mainly on protein sequences. However, it is not clear to what extent other important features, such as the symptoms caused by the viruses, could contribute to a predictor. Disease phenotypes (i.e. signs and symptoms) are readily accessible from clinical diagnosis and we hypothesize that they may act as a potential proxy and an additional source of information for the underlying molecular interactions between the pathogens and hosts.
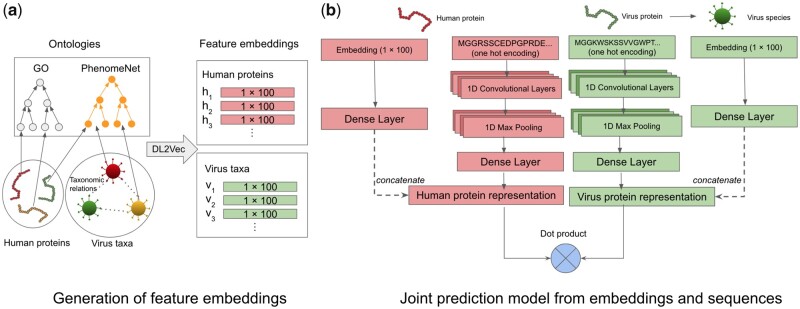
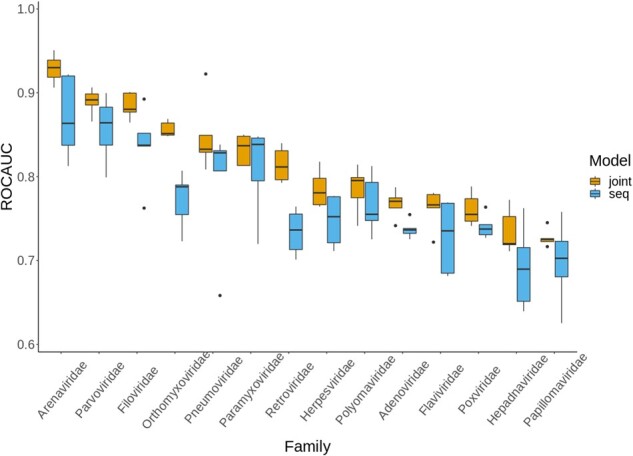
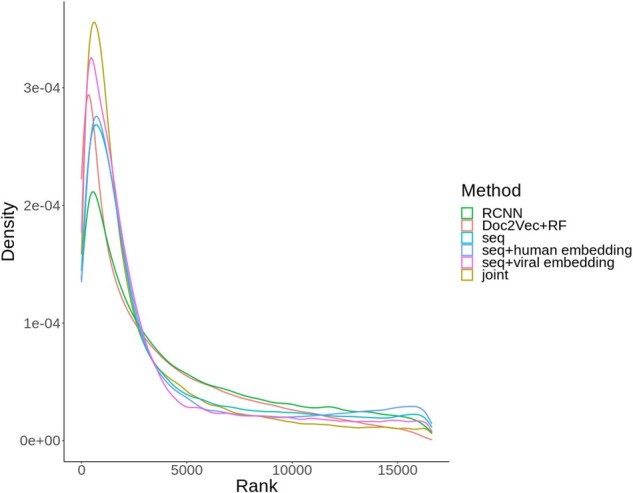
We developed DeepViral, a deep learning based method that predicts protein-protein interactions (PPI) between humans and viruses. Motivated by the potential utility of infectious disease phenotypes, we first embedded human proteins and viruses in a shared space using their associated phenotypes and functions, supported by formalized background knowledge from biomedical ontologies. By jointly learning from protein sequences and phenotype features, DeepViral significantly improves over existing sequence-based methods for intra- and inter-species PPI prediction.
Code and datasets for reproduction and customization are available at https://github.com/bio-ontology-research-group/DeepViral. Prediction results for 14 virus families are available at https://doi.org/10.5281/zenodo.4429824.
Supplementary data are available at Bioinformatics online.
新型病毒引起的传染病已成为主要的公共卫生问题。快速识别病毒与宿主的相互作用可以揭示传染病的机制,并为潜在治疗方法提供线索。目前针对新型病毒的计算预测方法主要基于蛋白质序列。然而,尚不清楚其他重要特征(如病毒引起的症状)在多大程度上有助于预测。疾病表型(即体征和症状)可从临床诊断中轻易获得,我们假设它们可能作为病原体与宿主之间潜在分子相互作用的潜在替代指标和额外信息来源。
我们开发了DeepViral,这是一种基于深度学习的方法,用于预测人类与病毒之间的蛋白质-蛋白质相互作用(PPI)。受传染病表型潜在效用的启发,我们首先利用生物医学本体的形式化背景知识,根据人类蛋白质和病毒的相关表型及功能,将它们嵌入到一个共享空间中。通过联合学习蛋白质序列和表型特征,DeepViral在种内和种间PPI预测方面比现有的基于序列的方法有显著改进。
补充数据可在《生物信息学》在线版获取。