Lee V Vern, Judd Louise M, Jex Aaron R, Holt Kathryn E, Tonkin Christopher J, Ralph Stuart A
Department of Biochemistry and Pharmacology, Bio21 Molecular Science and Biotechnology Institute, The University of Melbourne, Melbourne, Victoria, Australia.
The Walter and Eliza Hall Institute of Medical Research, Parkville, Melbourne, Victoria, Australia.
mSystems. 2021 Mar 9;6(2):e01081-20. doi: 10.1128/mSystems.01081-20.
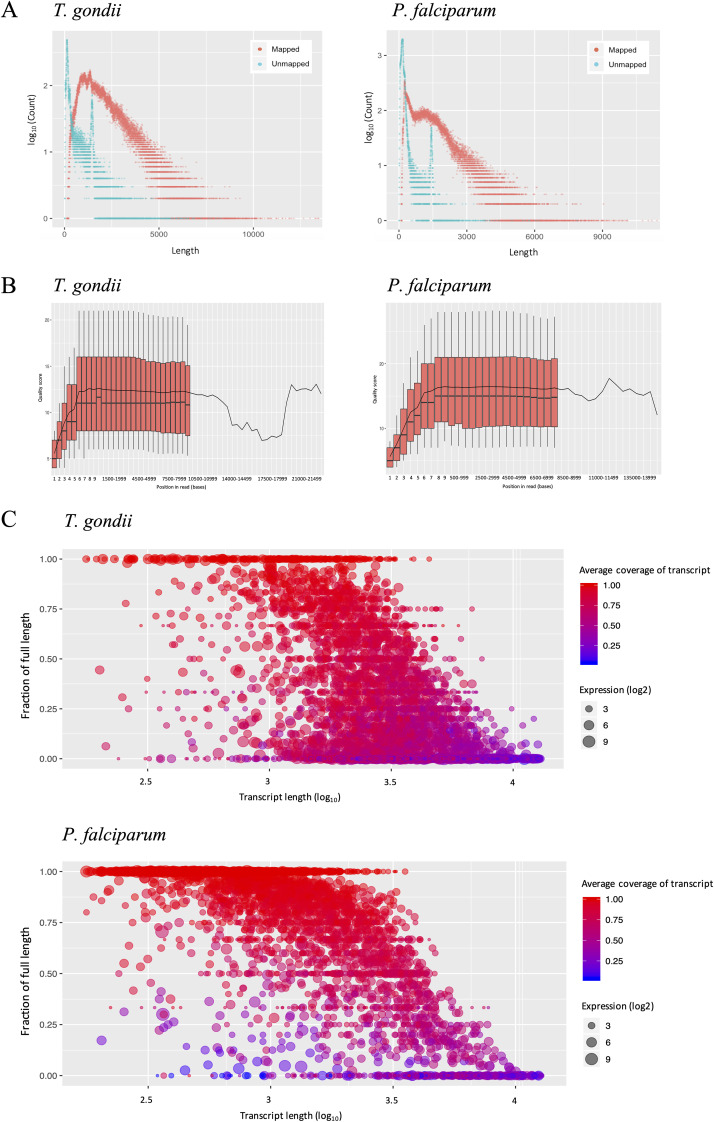
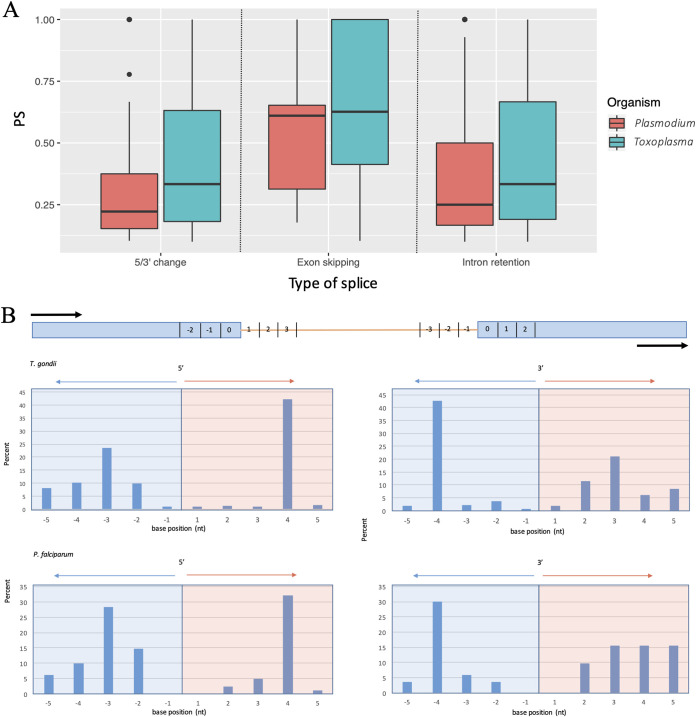
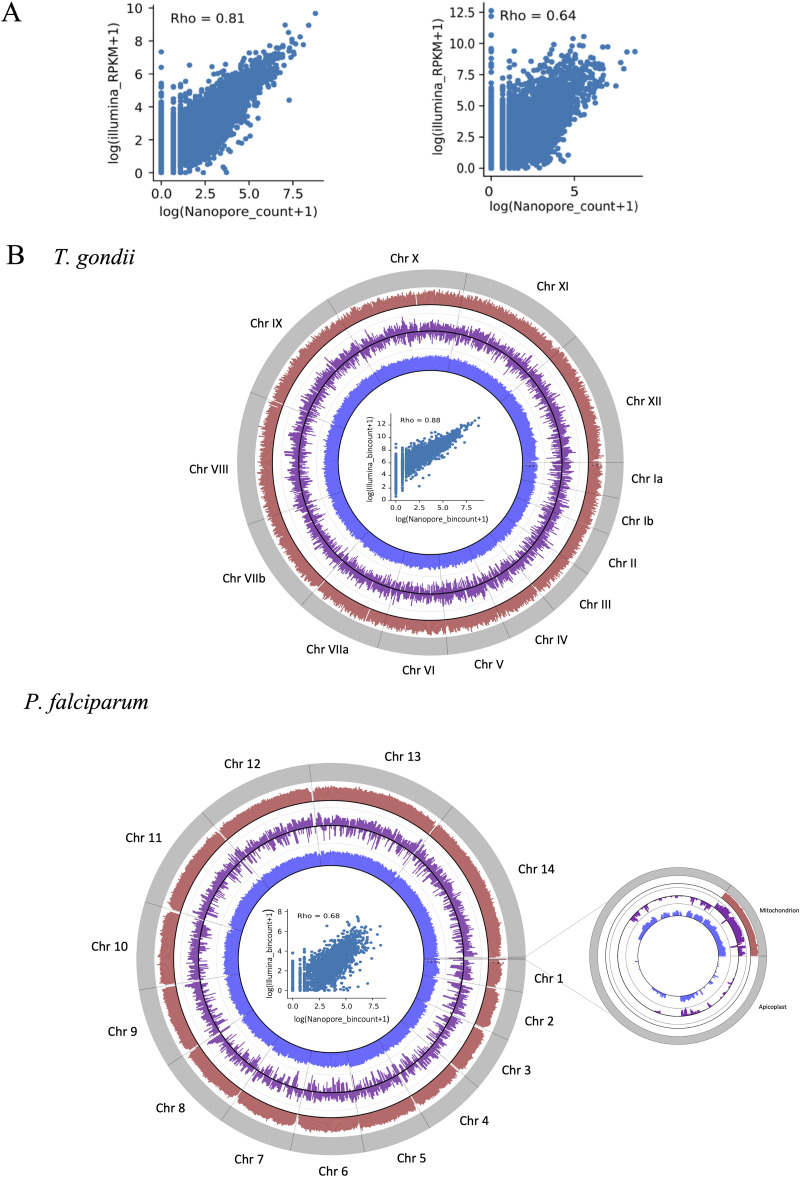
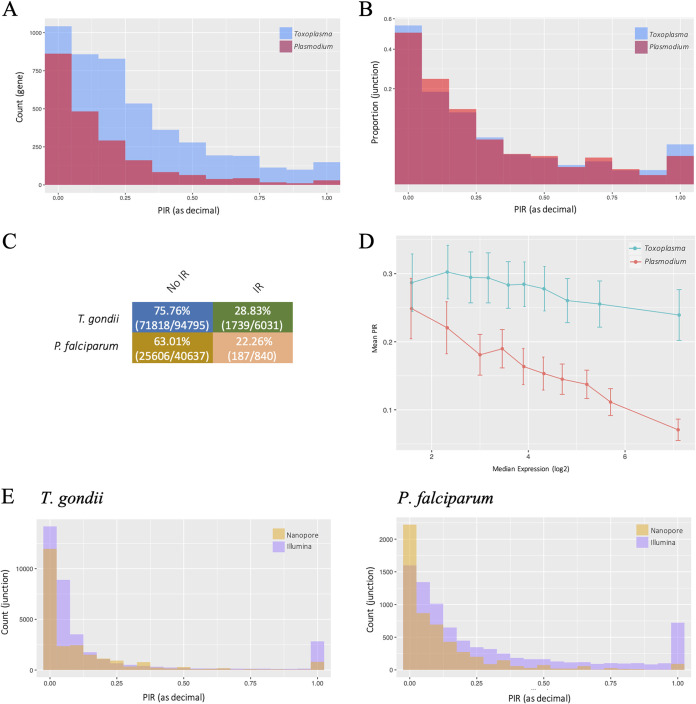
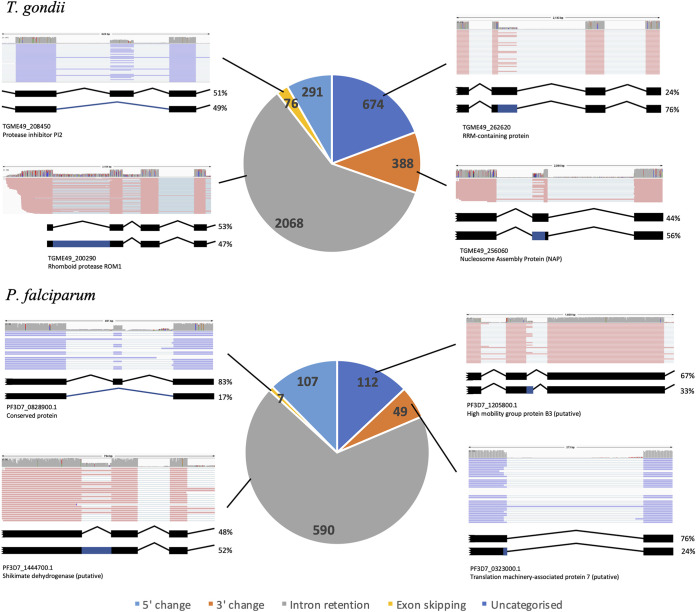
Alternative splicing is a widespread phenomenon in metazoans by which single genes are able to produce multiple isoforms of the gene product. However, this has been poorly characterized in apicomplexans, a major phylum of some of the most important global parasites. Efforts have been hampered by atypical transcriptomic features, such as the high AU content of RNA, but also the limitations of short-read sequencing in deciphering complex splicing events. In this study, we utilized the long read direct RNA sequencing platform developed by Oxford Nanopore Technologies to survey the alternative splicing landscape of and We find that while native RNA sequencing has a reduced throughput, it allows us to obtain full-length or nearly full-length transcripts with comparable quantification to Illumina sequencing. By comparing these data with available gene models, we find widespread alternative splicing, particularly intron retention, in these parasites. Most of these transcripts contain premature stop codons, suggesting that in these parasites, alternative splicing represents a pathway to transcriptomic diversity, rather than expanding proteomic diversity. Moreover, alternative splicing rates are comparable between parasites, suggesting a shared splicing machinery, despite notable transcriptomic differences between the parasites. This study highlights a strategy in using long-read sequencing to understand splicing events at the whole-transcript level and has implications in the future interpretation of transcriptome sequencing studies. We have used a novel nanopore sequencing technology to directly analyze parasite transcriptomes. The very long reads of this technology reveal the full-length genes of the parasites that cause malaria and toxoplasmosis. Gene transcripts must be processed in a process called splicing before they can be translated to protein. Our analysis reveals that these parasites very frequently only partially process their gene products, in a manner that departs dramatically from their human hosts.
可变剪接是后生动物中一种普遍存在的现象,通过这种现象单个基因能够产生多种基因产物的异构体。然而,在顶复门生物中,这种现象的特征却鲜为人知,顶复门是一些最重要的全球寄生虫的主要门类。研究工作受到非典型转录组特征的阻碍,比如RNA中高AU含量,同时短读长测序在解读复杂剪接事件方面也存在局限性。在本研究中,我们利用牛津纳米孔技术公司开发的长读长直接RNA测序平台来研究疟原虫和弓形虫的可变剪接情况。我们发现,虽然原生RNA测序通量较低,但它能让我们获得全长或近乎全长的转录本,其定量结果与Illumina测序相当。通过将这些数据与现有的基因模型进行比较,我们发现这些寄生虫中存在广泛的可变剪接现象,尤其是内含子保留。这些转录本中的大多数都含有提前终止密码子,这表明在这些寄生虫中,可变剪接代表了一种产生转录组多样性的途径,而非扩展蛋白质组多样性。此外,尽管这些寄生虫之间存在显著的转录组差异,但它们的可变剪接率相当,这表明存在共享的剪接机制。本研究突出了一种利用长读长测序来在全转录本水平理解剪接事件的策略,并对未来转录组测序研究的解读具有启示意义。我们使用了一种新型的纳米孔测序技术直接分析寄生虫转录组。这项技术的超长读长揭示了导致疟疾和弓形虫病的寄生虫的全长基因。基因转录本在能够被翻译成蛋白质之前,必须经过一个叫做剪接的过程进行加工。我们的分析表明,这些寄生虫非常频繁地仅对其基因产物进行部分加工,其方式与它们的人类宿主有很大不同。