Department of Biological Sciences-Marine and Environmental Biology, University of Southern California, Los Angeles, CA 90089
Department of Biological Sciences-Marine and Environmental Biology, University of Southern California, Los Angeles, CA 90089.
Proc Natl Acad Sci U S A. 2021 Mar 23;118(12). doi: 10.1073/pnas.2016810118.
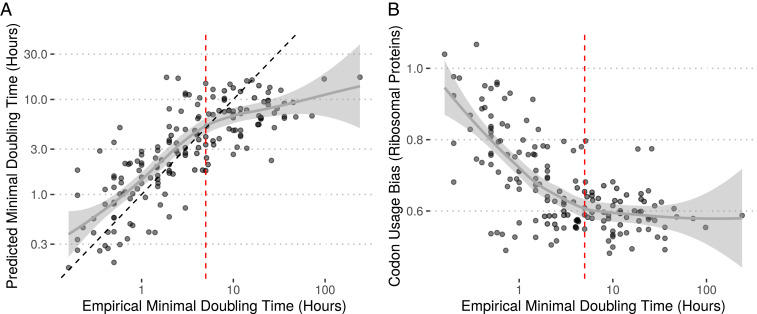
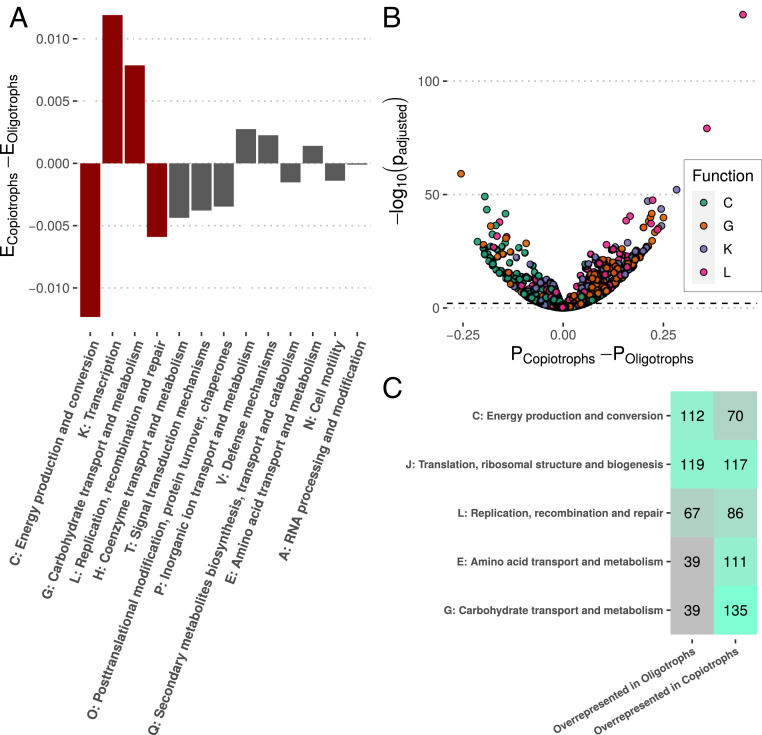
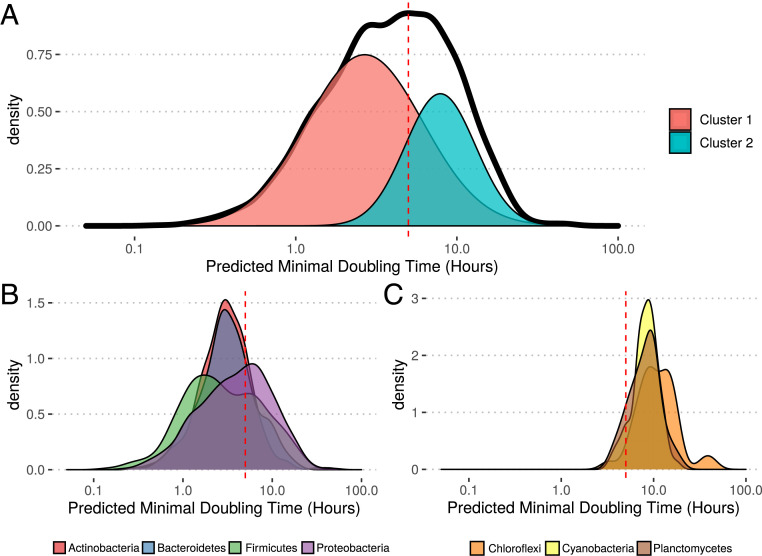
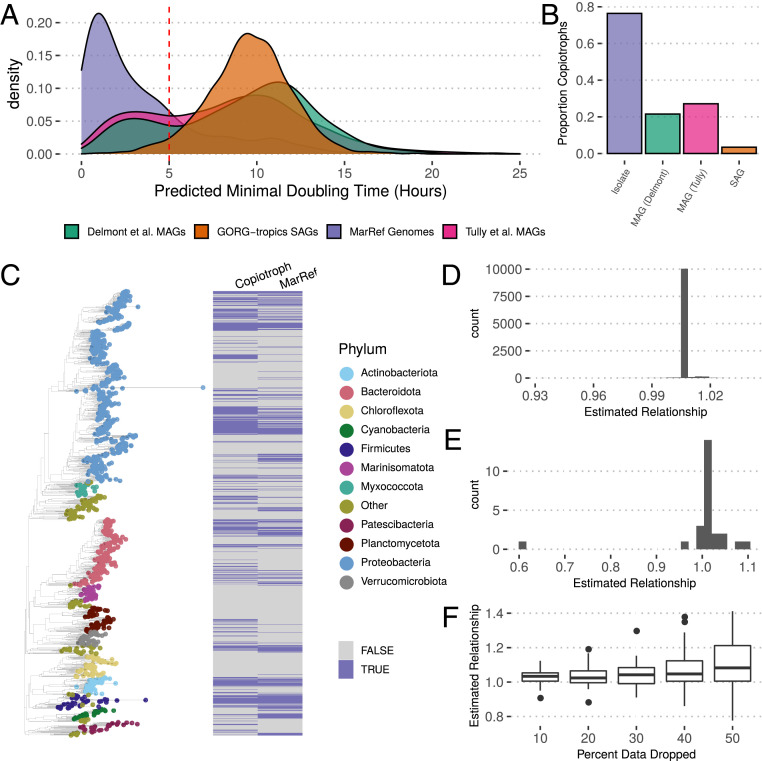
Maximal growth rate is a basic parameter of microbial lifestyle that varies over several orders of magnitude, with doubling times ranging from a matter of minutes to multiple days. Growth rates are typically measured using laboratory culture experiments. Yet, we lack sufficient understanding of the physiology of most microbes to design appropriate culture conditions for them, severely limiting our ability to assess the global diversity of microbial growth rates. Genomic estimators of maximal growth rate provide a practical solution to survey the distribution of microbial growth potential, regardless of cultivation status. We developed an improved maximal growth rate estimator and predicted maximal growth rates from over 200,000 genomes, metagenome-assembled genomes, and single-cell amplified genomes to survey growth potential across the range of prokaryotic diversity; extensions allow estimates from 16S rRNA sequences alone as well as weighted community estimates from metagenomes. We compared the growth rates of cultivated and uncultivated organisms to illustrate how culture collections are strongly biased toward organisms capable of rapid growth. Finally, we found that organisms naturally group into two growth classes and observed a bias in growth predictions for extremely slow-growing organisms. These observations ultimately led us to suggest evolutionary definitions of oligotrophy and copiotrophy based on the selective regime an organism occupies. We found that these growth classes are associated with distinct selective regimes and genomic functional potentials.
最大生长速率是微生物生活方式的一个基本参数,其变化幅度跨越几个数量级,倍增时间从几分钟到多天不等。生长速率通常通过实验室培养实验来测量。然而,我们对大多数微生物的生理学缺乏足够的了解,无法为它们设计合适的培养条件,这严重限制了我们评估微生物生长速率全球多样性的能力。最大生长速率的基因组估计值为调查微生物生长潜力的分布提供了一种实用的解决方案,无论培养状态如何。我们开发了一种改进的最大生长速率估计器,并从超过 20 万个基因组、宏基因组组装基因组和单细胞扩增基因组中预测了最大生长速率,以调查整个原核生物多样性范围内的生长潜力;扩展允许仅从 16S rRNA 序列以及从宏基因组中进行加权群落估计。我们比较了培养和未培养生物的生长速率,说明了培养物如何强烈偏向于能够快速生长的生物。最后,我们发现生物体自然分为两个生长类群,并观察到对生长极其缓慢的生物体的生长预测存在偏差。这些观察结果最终促使我们根据生物体所处的选择机制,提出了寡营养和富营养的进化定义。我们发现这些生长类群与不同的选择机制和基因组功能潜力相关。