School of Biotechnology and Biomolecular Sciences, University of New South Wales, Sydney, NSW, 2052, Australia.
Centre for Tropical Bioinformatics and Molecular Biology, Australian Institute of Tropical Health and Medicine, James Cook University, Cairns, QLD, 4878, Australia.
BMC Genomics. 2021 Mar 16;22(1):188. doi: 10.1186/s12864-021-07493-6.
Basenjis are considered an ancient dog breed of central African origins that still live and hunt with tribesmen in the African Congo. Nicknamed the barkless dog, Basenjis possess unique phylogeny, geographical origins and traits, making their genome structure of great interest. The increasing number of available canid reference genomes allows us to examine the impact the choice of reference genome makes with regard to reference genome quality and breed relatedness.
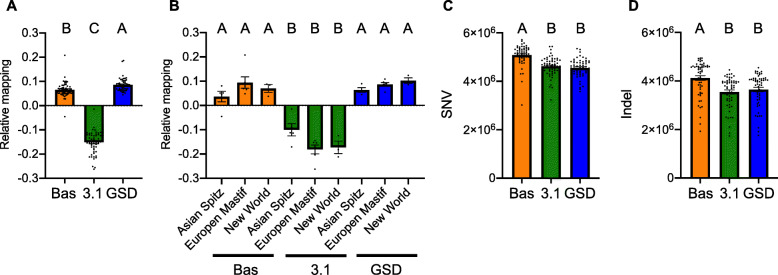
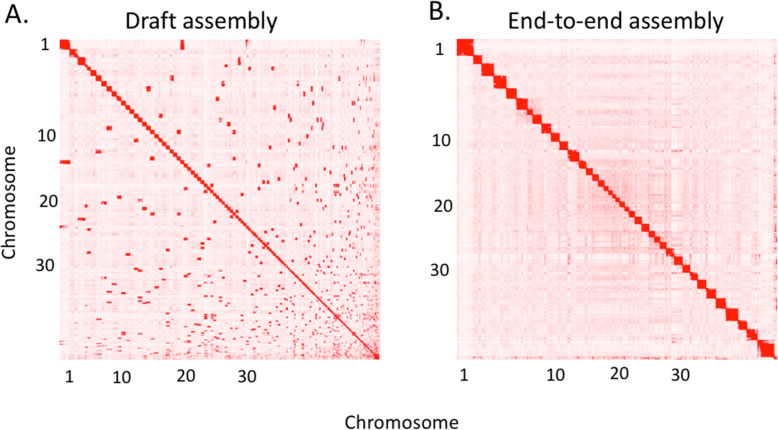
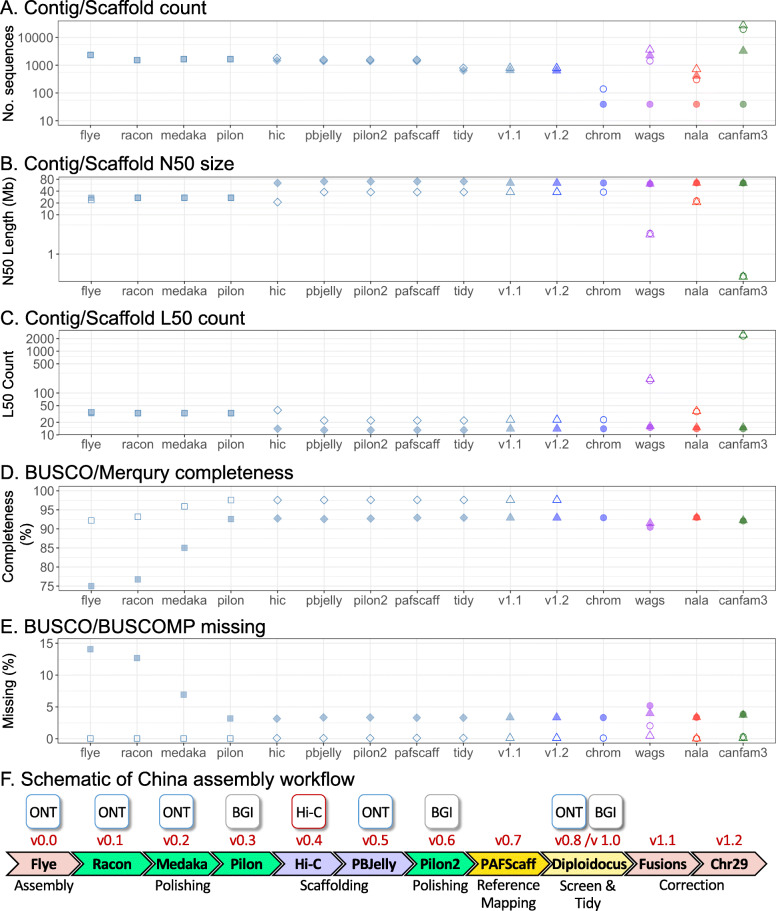
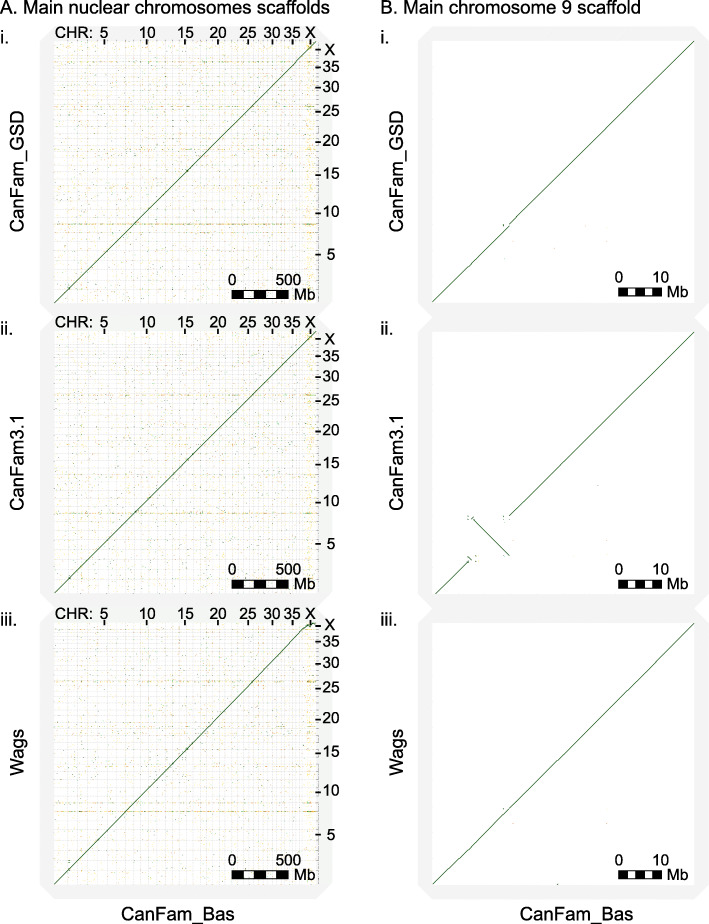
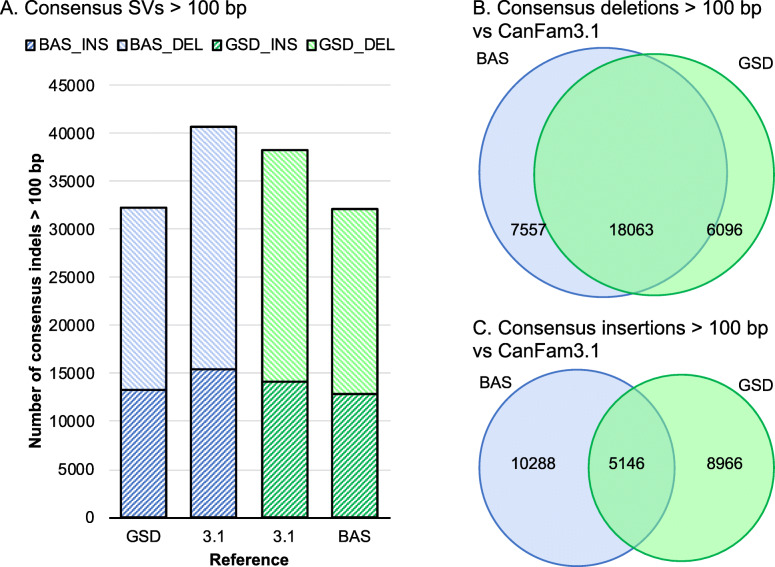
Here, we report two high quality de novo Basenji genome assemblies: a female, China (CanFam_Bas), and a male, Wags. We conduct pairwise comparisons and report structural variations between assembled genomes of three dog breeds: Basenji (CanFam_Bas), Boxer (CanFam3.1) and German Shepherd Dog (GSD) (CanFam_GSD). CanFam_Bas is superior to CanFam3.1 in terms of genome contiguity and comparable overall to the high quality CanFam_GSD assembly. By aligning short read data from 58 representative dog breeds to three reference genomes, we demonstrate how the choice of reference genome significantly impacts both read mapping and variant detection.
The growing number of high-quality canid reference genomes means the choice of reference genome is an increasingly critical decision in subsequent canid variant analyses. The basal position of the Basenji makes it suitable for variant analysis for targeted applications of specific dog breeds. However, we believe more comprehensive analyses across the entire family of canids is more suited to a pangenome approach. Collectively this work highlights the importance the choice of reference genome makes in all variation studies.
贝生吉犬被认为是一种古老的犬种,起源于中非,至今仍与非洲刚果的部落居民一起生活和狩猎。因其不会吠叫,贝生吉犬具有独特的系统发育、地理起源和特征,这使得它们的基因组结构备受关注。越来越多的犬类参考基因组可供使用,使我们能够研究参考基因组的选择对参考基因组质量和品种相关性的影响。
在这里,我们报告了两个高质量的贝生吉犬从头基因组组装:一个雌性,中国(CanFam_Bas),和一个雄性,Wags。我们进行了成对比较,并报告了三个犬种(贝生吉犬(CanFam_Bas)、拳师犬(CanFam3.1)和德国牧羊犬(CanFam_GSD))组装基因组之间的结构变异。CanFam_Bas 在基因组连续性方面优于 CanFam3.1,整体上与高质量的 CanFam_GSD 组装相当。通过将 58 个代表性犬种的短读序列数据与三个参考基因组进行比对,我们展示了参考基因组的选择如何显著影响读段比对和变异检测。
越来越多的高质量犬类参考基因组意味着参考基因组的选择在后续的犬类变异分析中是一个越来越关键的决策。贝生吉犬的基础位置使其适合于特定犬种的靶向应用的变异分析。然而,我们认为更全面的分析整个犬科家族更适合泛基因组方法。总的来说,这项工作强调了参考基因组选择在所有变异研究中的重要性。