Department of Veterinary Diagnostic and Production Animal Medicine, Iowa State University, Ames, Iowa, USA.
Bioinformatics and Computational Biology Program, Iowa State University, Ames, Iowa, USA.
mSphere. 2021 Mar 17;6(2):e00920-20. doi: 10.1128/mSphere.00920-20.
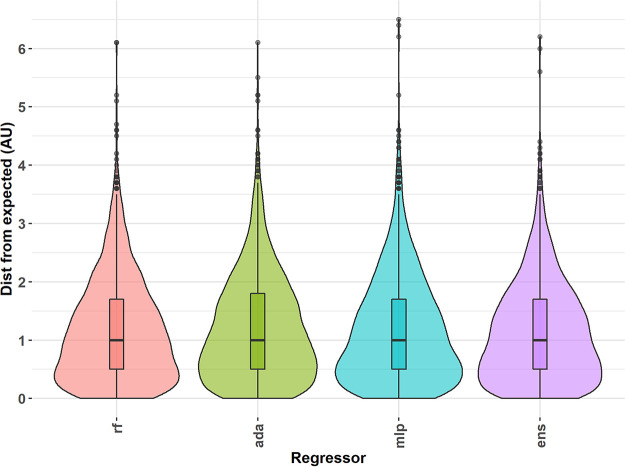
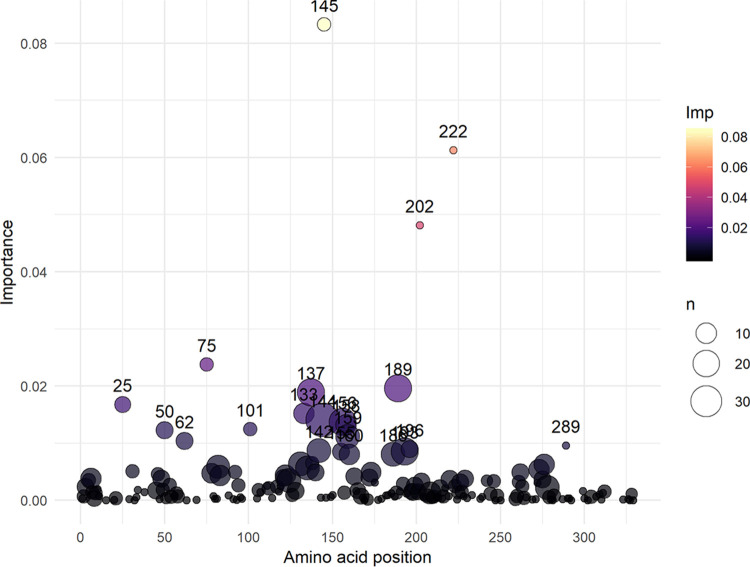
The antigenic diversity of influenza A viruses (IAV) circulating in swine challenges the development of effective vaccines, increasing zoonotic threat and pandemic potential. High-throughput sequencing technologies can quantify IAV genetic diversity, but there are no accurate approaches to adequately describe antigenic phenotypes. This study evaluated an ensemble of nonlinear regression models to estimate virus phenotype from genotype. Regression models were trained with a phenotypic data set of pairwise hemagglutination inhibition (HI) assays, using genetic sequence identity and pairwise amino acid mutations as predictor features. The model identified amino acid identity, ranked the relative importance of mutations in the hemagglutinin (HA) protein, and demonstrated good prediction accuracy. Four previously untested IAV strains were selected to experimentally validate model predictions by HI assays. Errors between predicted and measured distances of uncharacterized strains were 0.35, 0.61, 1.69, and 0.13 antigenic units. These empirically trained regression models can be used to estimate antigenic distances between different strains of IAV in swine by using sequence data. By ranking the importance of mutations in the HA, we provide criteria for identifying antigenically advanced IAV strains that may not be controlled by existing vaccines and can inform strain updates to vaccines to better control this pathogen. Influenza A viruses (IAV) in swine constitute a major economic burden to an important global agricultural sector, impact food security, and are a public health threat. Despite significant improvement in surveillance for IAV in swine over the past 10 years, sequence data have not been integrated into a systematic vaccine strain selection process for predicting antigenic phenotype and identifying determinants of antigenic drift. To overcome this, we developed nonlinear regression models that predict antigenic phenotype from genetic sequence data by training the model on hemagglutination inhibition assay results. We used these models to predict antigenic phenotype for previously uncharacterized IAV, ranked the importance of genetic features for antigenic phenotype, and experimentally validated our predictions. Our model predicted virus antigenic characteristics from genetic sequence data and provides a rapid and accurate method linking genetic sequence data to antigenic characteristics. This approach also provides support for public health by identifying viruses that are antigenically advanced from strains used as pandemic preparedness candidate vaccine viruses.
甲型流感病毒(IAV)在猪群中循环的抗原多样性给有效疫苗的开发带来了挑战,增加了人畜共患病的威胁和大流行的潜力。高通量测序技术可以量化 IAV 的遗传多样性,但目前还没有准确的方法来充分描述抗原表型。本研究评估了一组非线性回归模型,以从基因型估计病毒表型。使用遗传序列同一性和成对氨基酸突变作为预测特征,使用配对血凝抑制(HI)测定的表型数据集对回归模型进行了训练。该模型确定了氨基酸同一性,对血凝素(HA)蛋白中的突变进行了相对重要性排序,并展示了良好的预测准确性。选择了四个以前未测试的 IAV 株,通过 HI 测定实验验证模型预测。未表征菌株的预测和测量距离之间的误差为 0.35、0.61、1.69 和 0.13 个抗原单位。这些经验训练的回归模型可以通过使用序列数据来估计猪中不同 IAV 株之间的抗原距离。通过对 HA 中突变的重要性进行排序,我们提供了识别可能不受现有疫苗控制的抗原先进 IAV 株的标准,并为疫苗更新提供信息,以更好地控制这种病原体。猪中的甲型流感病毒(IAV)对全球重要的农业部门构成了重大的经济负担,影响了粮食安全,也是公共卫生威胁。尽管在过去 10 年中对猪流感病毒的监测有了显著改善,但序列数据尚未整合到系统的疫苗株选择过程中,以预测抗原表型并确定抗原漂移的决定因素。为了克服这一问题,我们开发了非线性回归模型,通过在血凝抑制测定结果上训练模型,从遗传序列数据预测抗原表型。我们使用这些模型预测以前未表征的 IAV 的抗原表型,对遗传特征对抗原表型的重要性进行了排序,并对我们的预测进行了实验验证。我们的模型从遗传序列数据预测病毒的抗原特征,并提供了一种快速准确的方法,将遗传序列数据与抗原特征联系起来。这种方法还通过识别具有先进抗原性的病毒,为公共卫生提供了支持,这些病毒与作为大流行准备候选疫苗病毒的菌株不同。