School of Health and Life Sciences, Teesside University, Middlesbrough, TS4 3BX, UK.
School of Computing, Eng. & Digital Tech., Teesside University, Middlesbrough, TS4 3BX, UK.
Sci Rep. 2021 Mar 18;11(1):6265. doi: 10.1038/s41598-021-85285-4.
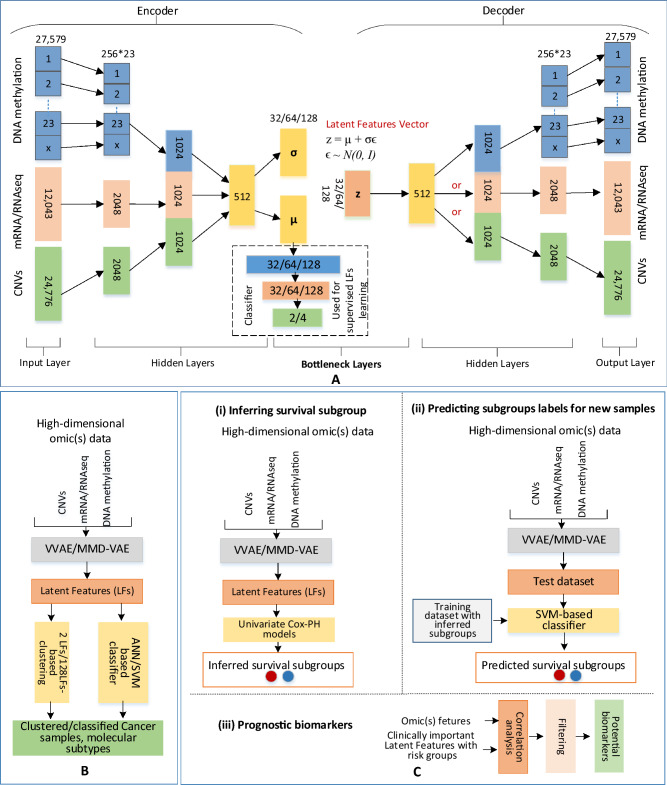
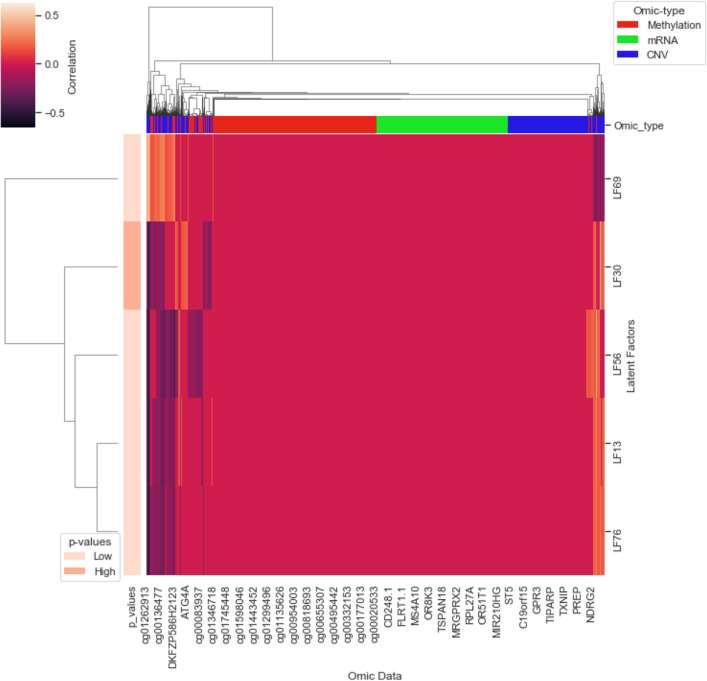
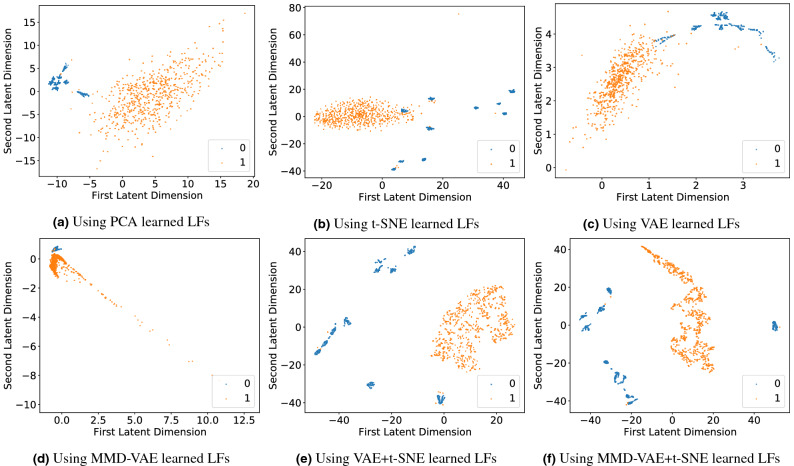
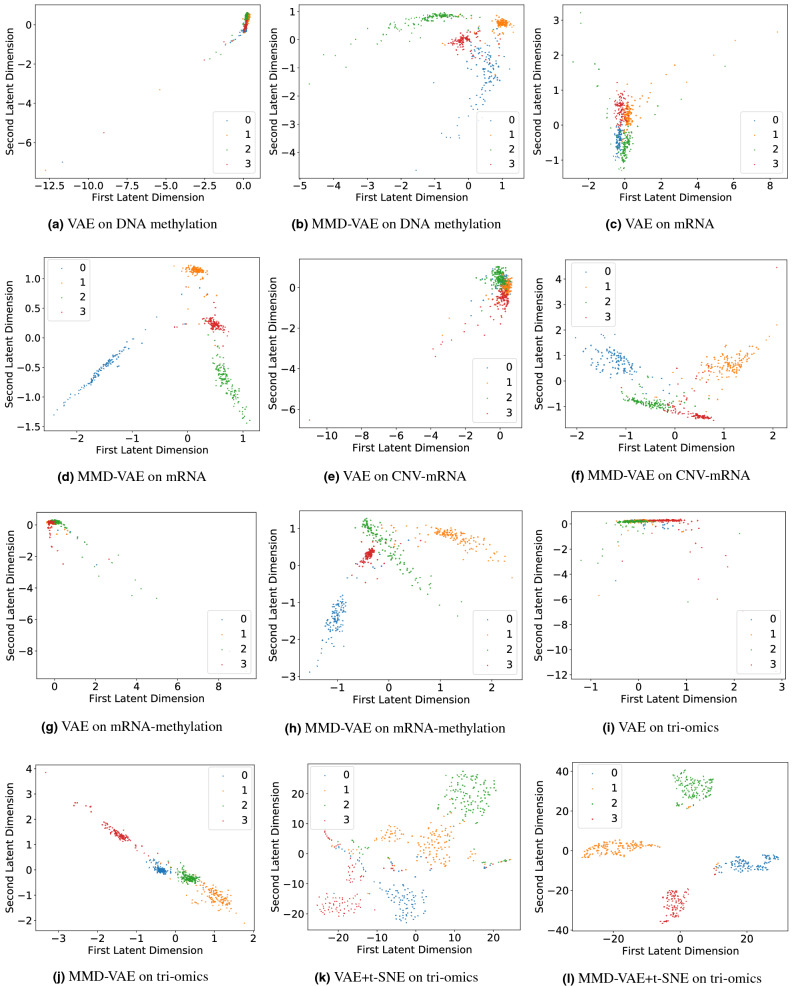
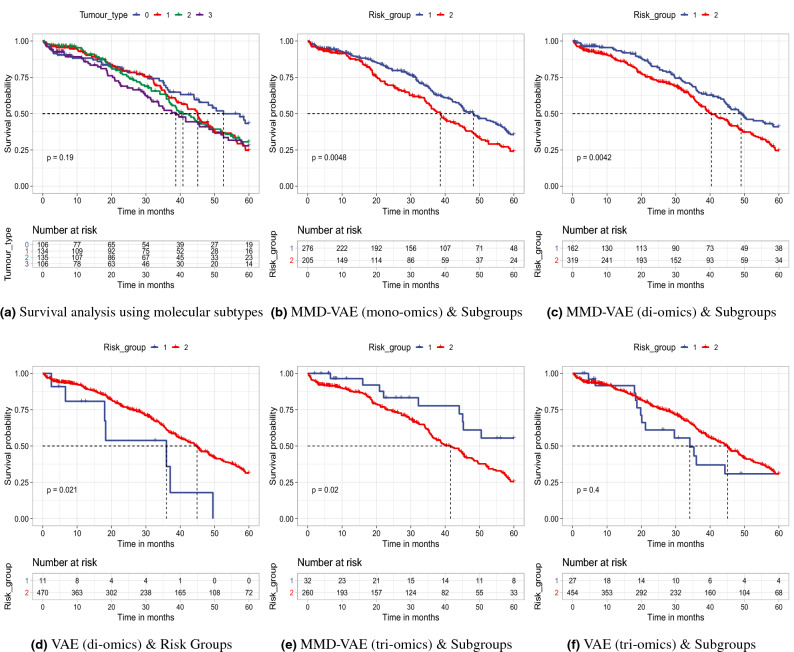
Cancer is a complex disease that deregulates cellular functions at various molecular levels (e.g., DNA, RNA, and proteins). Integrated multi-omics analysis of data from these levels is necessary to understand the aberrant cellular functions accountable for cancer and its development. In recent years, Deep Learning (DL) approaches have become a useful tool in integrated multi-omics analysis of cancer data. However, high dimensional multi-omics data are generally imbalanced with too many molecular features and relatively few patient samples. This imbalance makes a DL based integrated multi-omics analysis difficult. DL-based dimensionality reduction technique, including variational autoencoder (VAE), is a potential solution to balance high dimensional multi-omics data. However, there are few VAE-based integrated multi-omics analyses, and they are limited to pancancer. In this work, we did an integrated multi-omics analysis of ovarian cancer using the compressed features learned through VAE and an improved version of VAE, namely Maximum Mean Discrepancy VAE (MMD-VAE). First, we designed and developed a DL architecture for VAE and MMD-VAE. Then we used the architecture for mono-omics, integrated di-omics and tri-omics data analysis of ovarian cancer through cancer samples identification, molecular subtypes clustering and classification, and survival analysis. The results show that MMD-VAE and VAE-based compressed features can respectively classify the transcriptional subtypes of the TCGA datasets with an accuracy in the range of 93.2-95.5% and 87.1-95.7%. Also, survival analysis results show that VAE and MMD-VAE based compressed representation of omics data can be used in cancer prognosis. Based on the results, we can conclude that (i) VAE and MMD-VAE outperform existing dimensionality reduction techniques, (ii) integrated multi-omics analyses perform better or similar compared to their mono-omics counterparts, and (iii) MMD-VAE performs better than VAE in most omics dataset.
癌症是一种复杂的疾病,会在各种分子水平上(例如 DNA、RNA 和蛋白质)扰乱细胞功能。为了了解导致癌症及其发展的异常细胞功能,有必要对这些水平的数据进行整合的多组学分析。近年来,深度学习(DL)方法已成为癌症数据整合多组学分析的有用工具。然而,高维多组学数据通常存在不平衡问题,即有太多的分子特征和相对较少的患者样本。这种不平衡使得基于 DL 的整合多组学分析变得困难。基于 DL 的降维技术,包括变分自编码器(VAE),是平衡高维多组学数据的潜在解决方案。然而,基于 VAE 的整合多组学分析很少,并且仅限于泛癌。在这项工作中,我们使用通过 VAE 和 VAE 的改进版本最大均值差异变分自编码器(MMD-VAE)学习的压缩特征对卵巢癌进行了整合的多组学分析。首先,我们设计并开发了用于 VAE 和 MMD-VAE 的 DL 架构。然后,我们通过癌症样本识别、分子亚型聚类和分类以及生存分析,使用该架构对卵巢癌的单组学、整合的二组学和三组学数据进行分析。结果表明,MMD-VAE 和基于 VAE 的压缩特征可分别以 93.2-95.5%和 87.1-95.7%的范围内对 TCGA 数据集的转录亚型进行分类。此外,生存分析结果表明,基于 VAE 和 MMD-VAE 的组学数据的压缩表示可用于癌症预后。基于这些结果,我们可以得出结论:(i)VAE 和 MMD-VAE 优于现有的降维技术;(ii)整合的多组学分析比单组学分析表现更好或相似;(iii)在大多数组学数据集中,MMD-VAE 比 VAE 表现更好。