Wojtkiewicz Melinda, Berg Luecke Linda, Kelly Maia I, Gundry Rebekah L
CardiOmics Program, Center for Heart and Vascular Research; Division of Cardiovascular Medicine; and Department of Cellular and Integrative Physiology, University of Nebraska Medical Center, Omaha, Nebraska.
Department of Biochemistry, Medical College of Wisconsin, Milwaukee, Wisconsin.
Curr Protoc. 2021 Mar;1(3):e85. doi: 10.1002/cpz1.85.
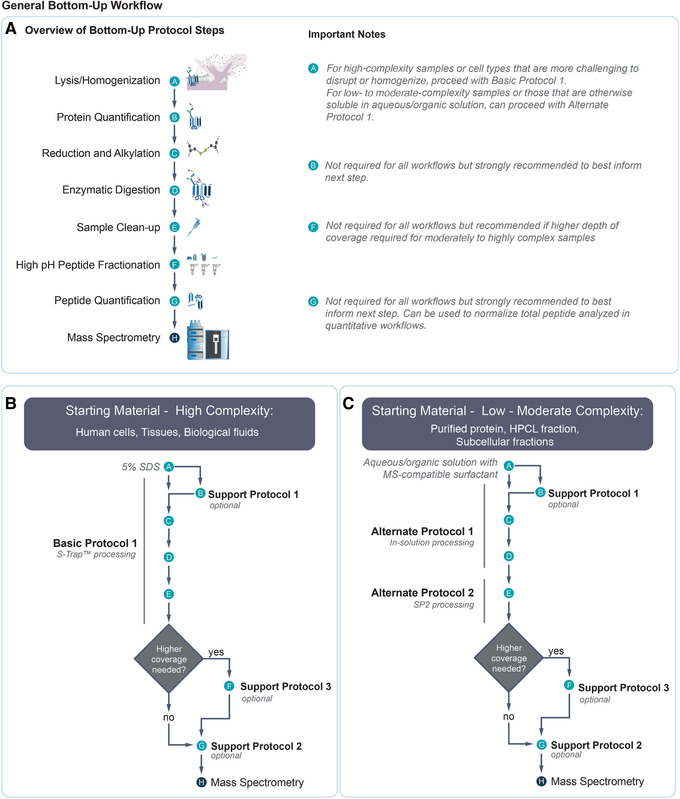
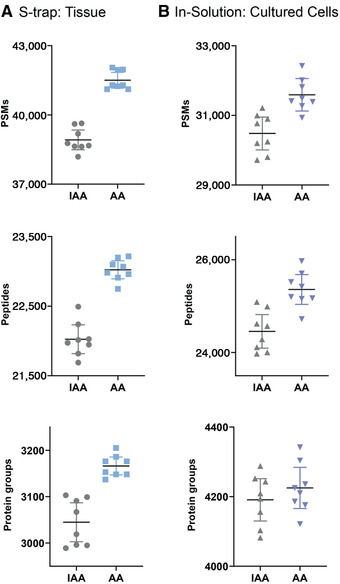
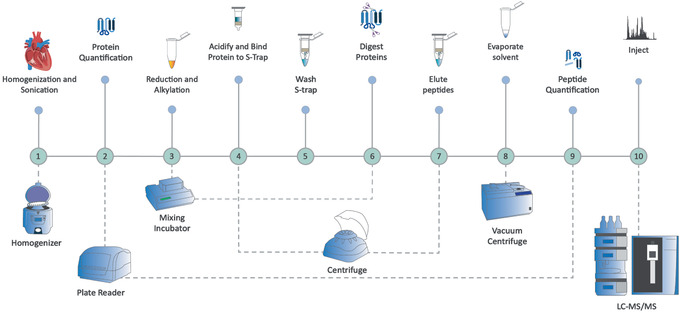
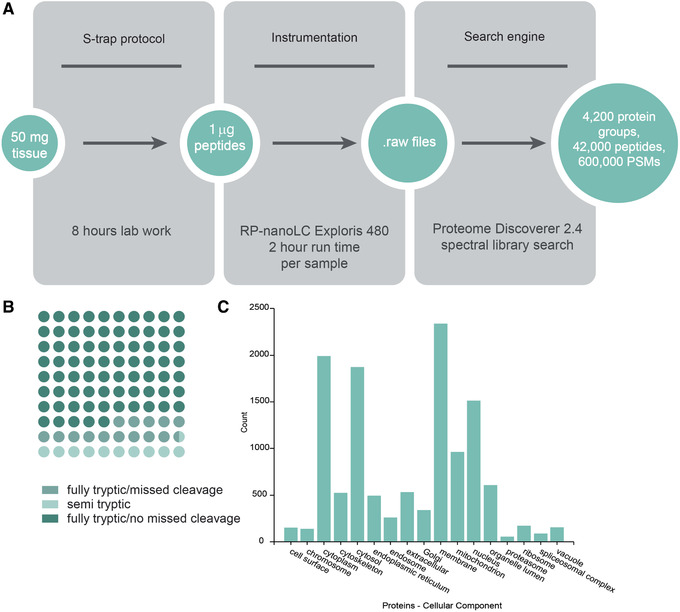
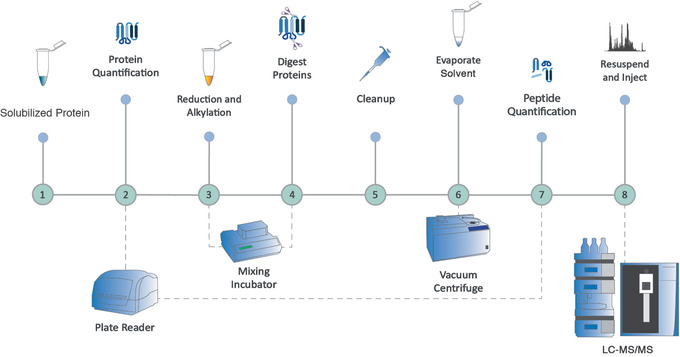
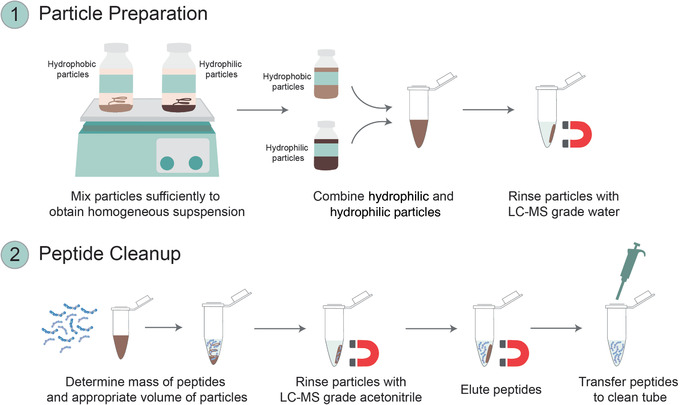
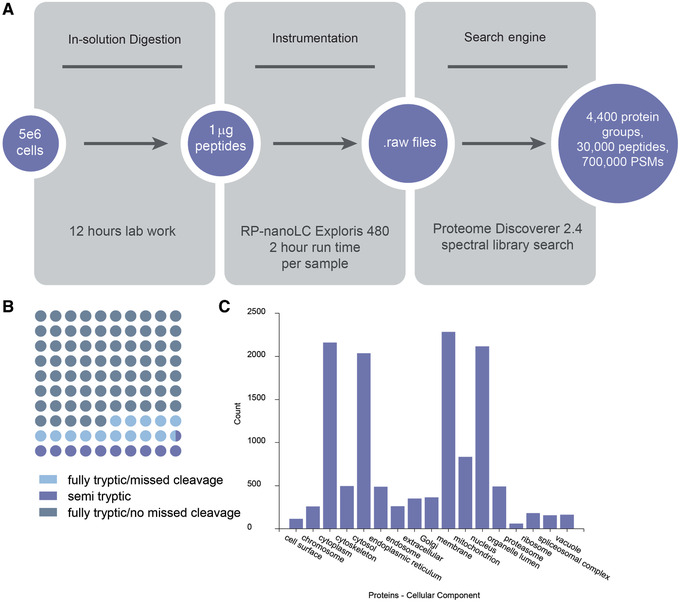
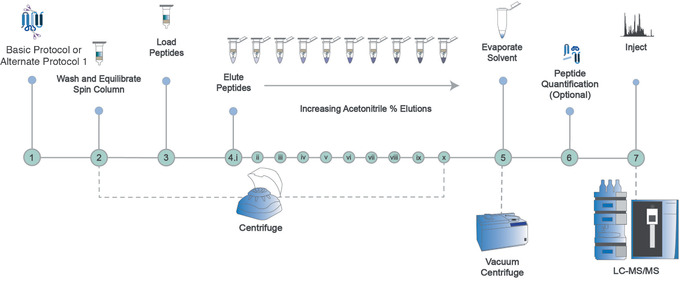
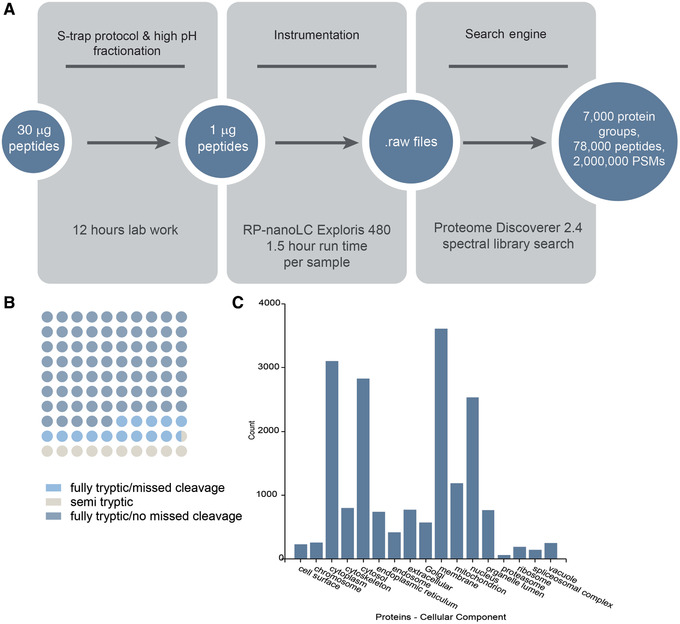
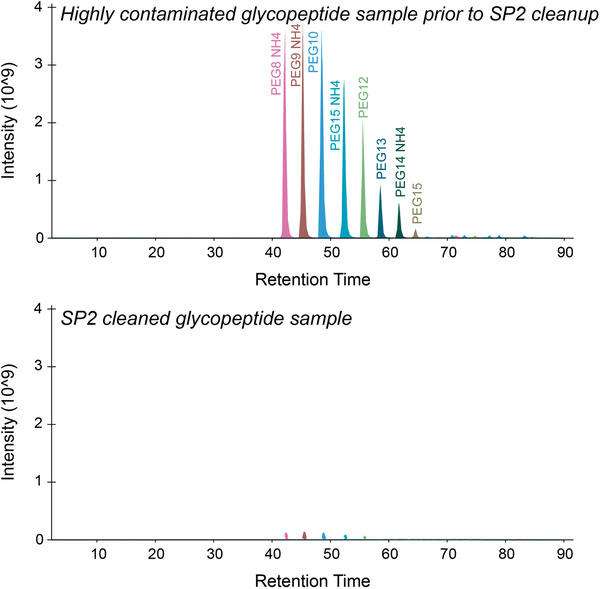
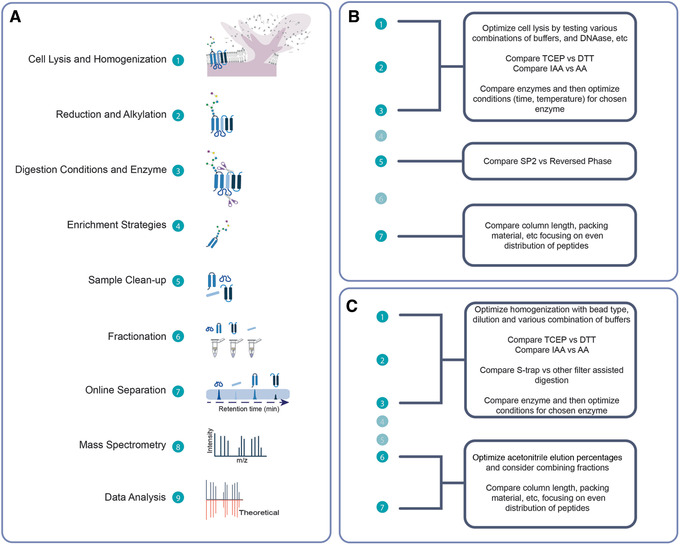
Mass spectrometry (MS) is routinely used to identify, characterize, and quantify biological molecules. For protein analysis, MS-based workflows can be broadly categorized as top-down or bottom-up, depending on whether the proteins are analyzed as intact molecules or first digested into peptides. This article outlines steps for preparing peptide samples for MS as part of a bottom-up proteomics workflow, providing versatile methods suitable for discovery and targeted analyses in qualitative and quantitative workflows. Resulting samples contain peptides of suitable size for analysis by MS instrumentation generally available to modern research laboratories, including MS coupled to either liquid chromatography (LC) or matrix-assisted laser desorption/ionization (MALDI) interfaces. This article incorporates recent developments in methodologies and consumables to facilitate sample preparation. The protocols are well-suited to users without prior experience in proteomics and include methods for universally applicable suspension trap processing and for alternate in-solution processing to accommodate a range of sample types. Cleanup, quantification, and fractionation procedures are also described. © 2021 The Authors. Basic Protocol: Preparation of high-complexity peptide samples for mass spectrometry analysis using S-Trap™ processing Alternate Protocol 1: Preparation of low- to moderate-complexity peptide samples for mass spectrometry analysis using in-solution processing Alternate Protocol 2: Detergent, polymer, and salt removal from peptide samples before mass spectrometry analysis using SP2 processing Support Protocol 1: Protein quantification using Pierce 660 nm assay Support Protocol 2: Peptide quantification using Pierce quantitative fluorometric peptide assay Support Protocol 3: High-pH fractionation of complex peptide samples.
质谱分析法(MS)通常用于鉴定、表征和定量生物分子。对于蛋白质分析,基于质谱的工作流程大致可分为自上而下或自下而上两种类型,这取决于蛋白质是作为完整分子进行分析,还是先被消化成肽段。本文概述了作为自下而上蛋白质组学工作流程一部分的用于质谱分析的肽样品制备步骤,提供了适用于定性和定量工作流程中发现性分析和靶向分析的通用方法。所得样品包含大小合适的肽段,可通过现代研究实验室普遍配备的质谱仪器进行分析,包括与液相色谱(LC)或基质辅助激光解吸/电离(MALDI)接口联用的质谱仪。本文纳入了方法和耗材方面的最新进展,以促进样品制备。这些方案非常适合没有蛋白质组学经验的用户,包括普遍适用的悬浮阱处理方法以及用于替代溶液内处理的方法,以适应一系列样品类型。还描述了净化、定量和分级分离程序。© 2021作者。基本方案:使用S-Trap™ 处理制备用于质谱分析的高复杂性肽样品替代方案1:使用溶液内处理制备用于质谱分析的低至中等复杂性肽样品替代方案2:在质谱分析前使用SP2处理从肽样品中去除去污剂、聚合物和盐分支持方案1:使用Pierce 660 nm检测法进行蛋白质定量支持方案2:使用Pierce定量荧光肽检测法进行肽定量支持方案3:复杂肽样品的高pH分级分离