Division of Cardiology, Department of Medicine, Brigham and Women's Hospital, Harvard Medical School, 75 Francis Street, Boston, MA 02115, USA.
TIMI Study Group, Boston, MA, USA.
Eur Heart J. 2021 May 21;42(20):1988-1996. doi: 10.1093/eurheartj/ehab148.
Childhood-onset hypertrophic cardiomyopathy (HCM) is far less common than adult-onset disease, thus natural history is not well characterized. We aim to describe the characteristics and outcomes of childhood-onset HCM.
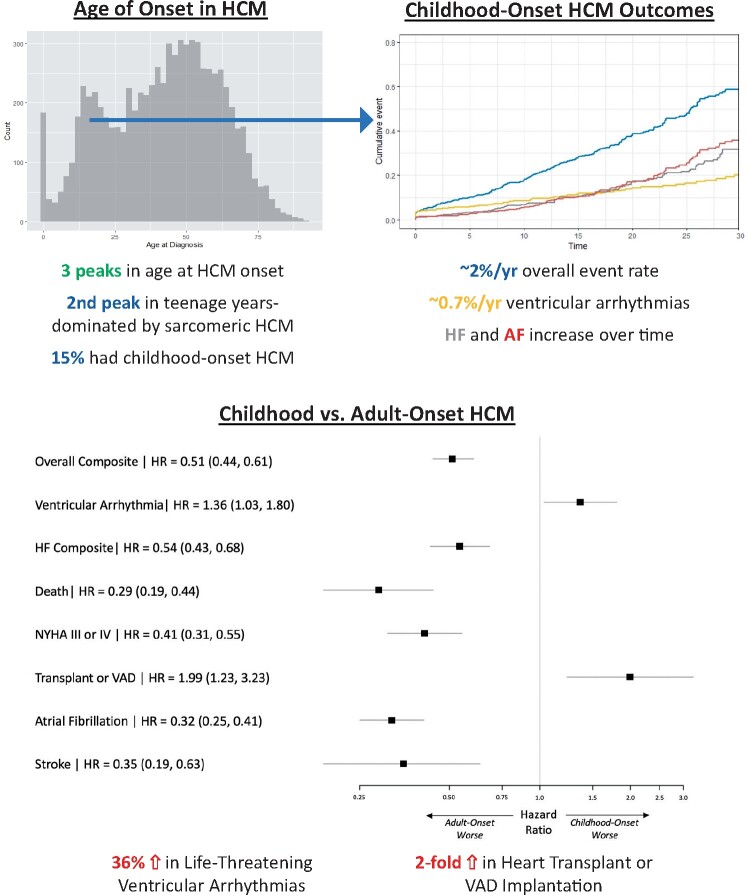
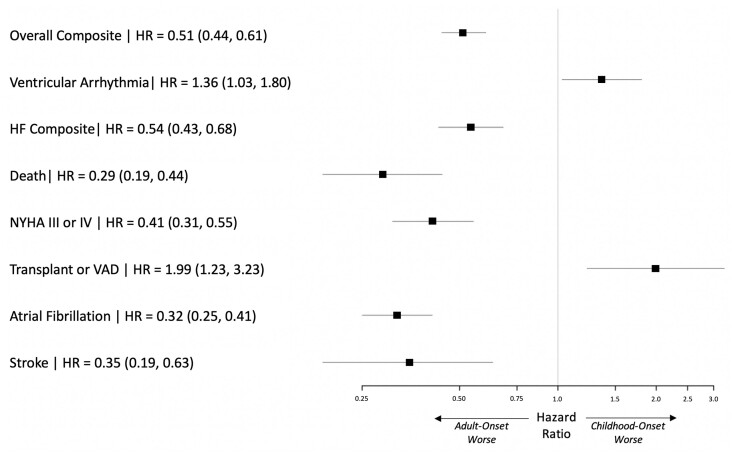
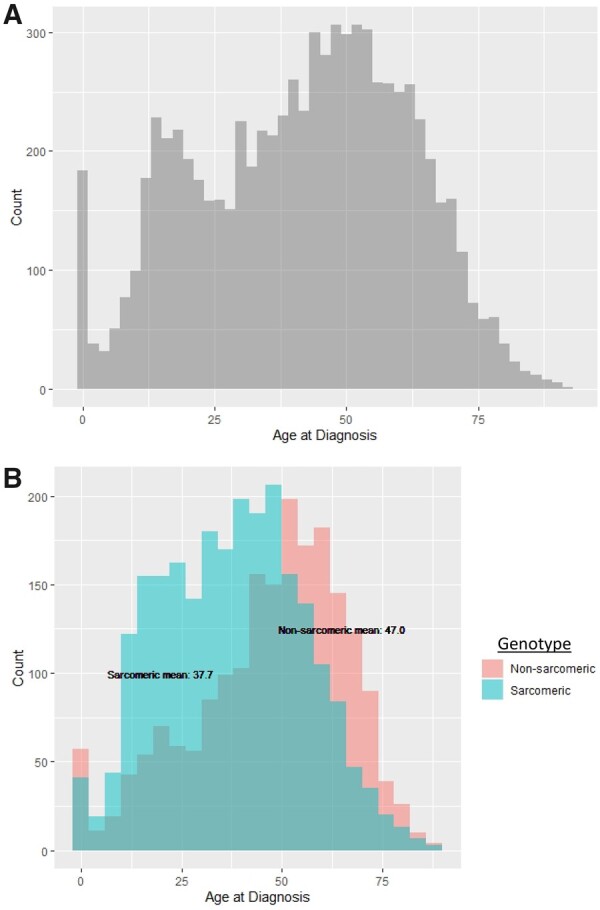
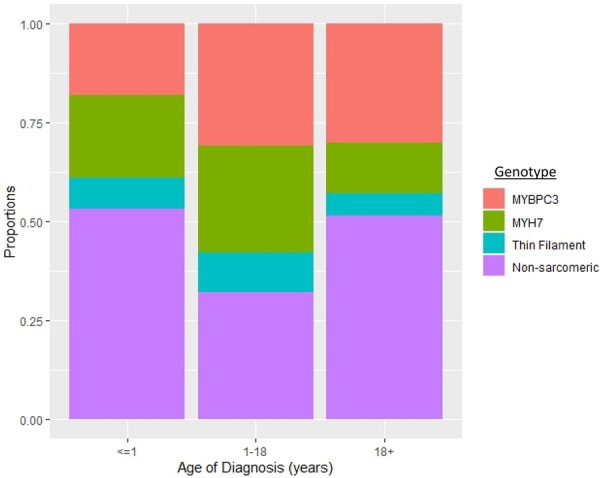
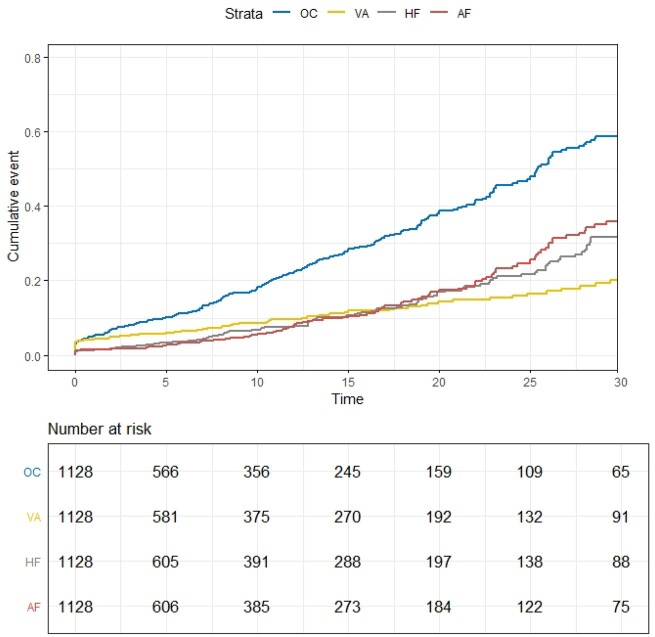
We performed an observational cohort study of 7677 HCM patients from the Sarcomeric Human Cardiomyopathy Registry (SHaRe). Hypertrophic cardiomyopathy patients were stratified by age at diagnosis [<1 year (infancy), 1-18 years (childhood), >18 years (adulthood)] and assessed for composite endpoints reflecting heart failure (HF), life-threatening ventricular arrhythmias, atrial fibrillation (AF), and an overall composite that also included stroke and death. Stratifying by age of diagnosis, 184 (2.4%) patients were diagnosed in infancy; 1128 (14.7%) in childhood; and 6365 (82.9%) in adulthood. Childhood-onset HCM patients had an ∼2%/year event rate for the overall composite endpoint, with ventricular arrhythmias representing the most common event in the 1st decade following baseline visit, but HF and AF becoming more common by the end of the 2nd decade. Sarcomeric variants were more common in childhood-onset HCM (63%) and carried a worse prognosis than non-sarcomeric disease, including a greater than two-fold increased risk of HF [HRadj 2.39 (1.36-4.20), P = 0.003] and 67% increased risk of the overall composite outcome [HRadj 1.67 (1.16-2.41), P = 0.006]. When compared with adult-onset HCM, childhood-onset was 36% more likely to develop life-threatening ventricular arrhythmias [HRadj 1.36 (1.03-1.80)] and twice as likely to require transplant or ventricular assist device [HRadj 1.99 (1.23-3.23)].
Patients with childhood-onset HCM are more likely to have sarcomeric disease, carry a higher risk of life-threatening ventricular arrythmias, and have greater need for advanced HF therapies. These findings provide insight into the natural history of disease and can help inform clinical risk stratification.
儿童起病型肥厚型心肌病(HCM)比成人起病型更为少见,因此其自然病史尚不清楚。本研究旨在描述儿童起病型 HCM 的特征和结局。
我们对 Sarcomeric Human Cardiomyopathy Registry(SHaRe)中 7677 例 HCM 患者进行了一项观察性队列研究。根据诊断时的年龄(<1 岁[婴儿期]、1-18 岁[儿童期]、>18 岁[成年期])将 HCM 患者分层,并评估反映心力衰竭(HF)、危及生命的室性心律失常、心房颤动(AF)以及同时包括卒中和死亡的整体复合终点的复合终点。按诊断年龄分层,184(2.4%)例患者在婴儿期被诊断,1128(14.7%)例在儿童期,6365(82.9%)例在成年期。儿童起病型 HCM 的整体复合终点事件发生率约为 2%/年,在基线后第 1 个 10 年,室性心律失常是最常见的事件,但在第 2 个 10 年末 HF 和 AF 更为常见。儿童起病型 HCM 中肌节变异更为常见(63%),预后较非肌节疾病差,HF 的风险增加两倍以上[HRadj 2.39(1.36-4.20),P=0.003],整体复合结局的风险增加 67%[HRadj 1.67(1.16-2.41),P=0.006]。与成年起病型 HCM 相比,儿童起病型更有可能发生危及生命的室性心律失常[HRadj 1.36(1.03-1.80)],需要进行心脏移植或心室辅助装置治疗的可能性增加一倍[HRadj 1.99(1.23-3.23)]。
儿童起病型 HCM 患者更有可能患肌节疾病,发生危及生命的室性心律失常的风险更高,且更需要先进的 HF 治疗。这些发现为疾病的自然病史提供了深入了解,并有助于指导临床风险分层。