Burnett School of Biomedical Sciences, Genomics and Bioinformatics Cluster, University of Central Florida, Orlando, 32816, FL, USA.
Department of Computer Science, Genomics and Bioinformatics Cluster, University of Central Florida, Orlando, FL, 32816, USA.
BMC Microbiol. 2021 Mar 31;21(1):98. doi: 10.1186/s12866-021-02153-x.
Colorectal cancer is a leading cause of cancer-related deaths worldwide. The human gut microbiome has become an active area of research for understanding the initiation, progression, and treatment of colorectal cancer. Despite multiple studies having found significant alterations in the carriage of specific bacteria within the gut microbiome of colorectal cancer patients, no single bacterium has been unequivocally connected to all cases. Whether alterations in species carriages are the cause or outcome of cancer formation is still unclear, but what is clear is that focus should be placed on understanding changes to the bacterial community structure within the cancer-associated gut microbiome.
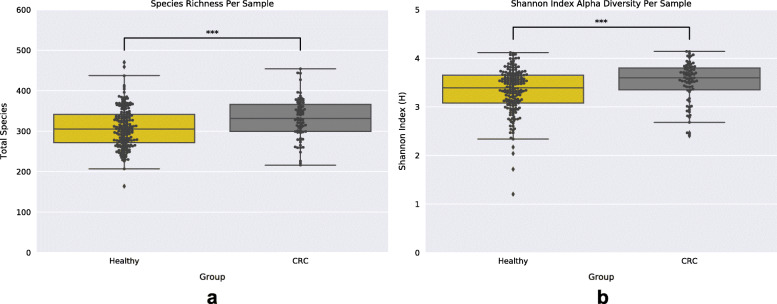
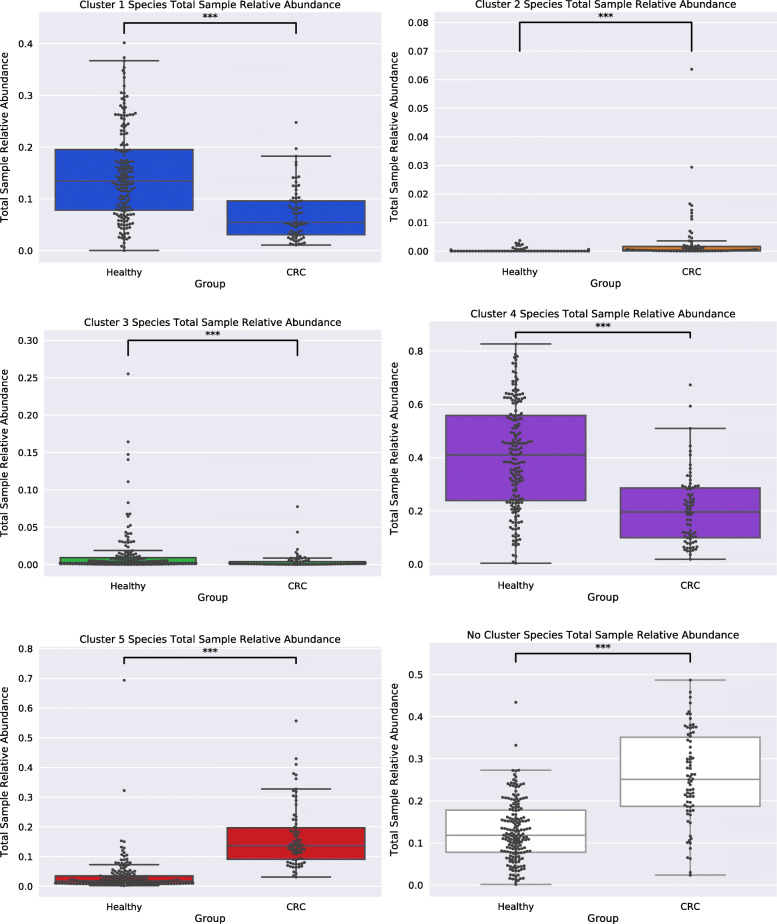
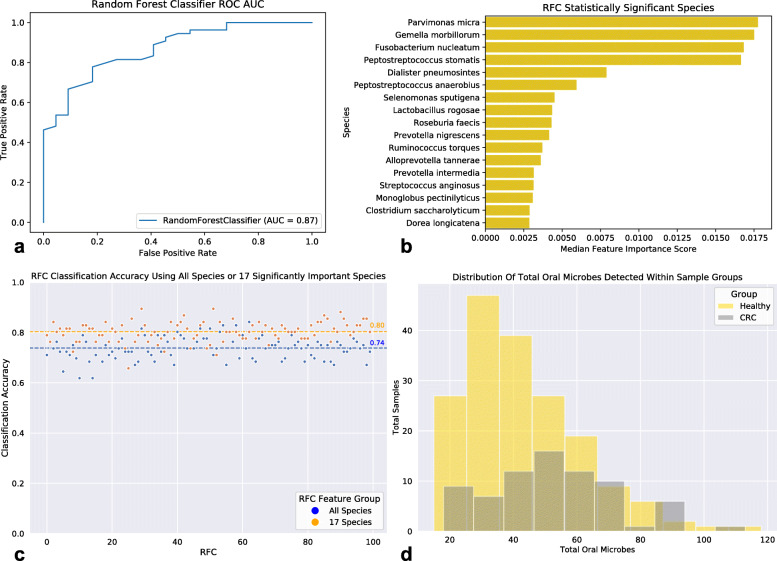
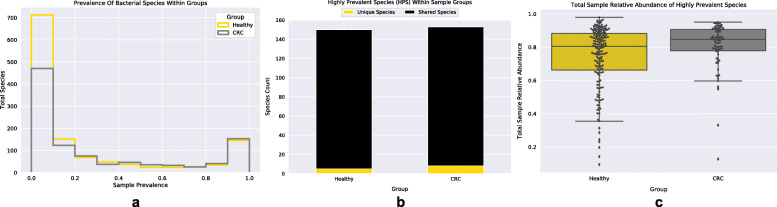
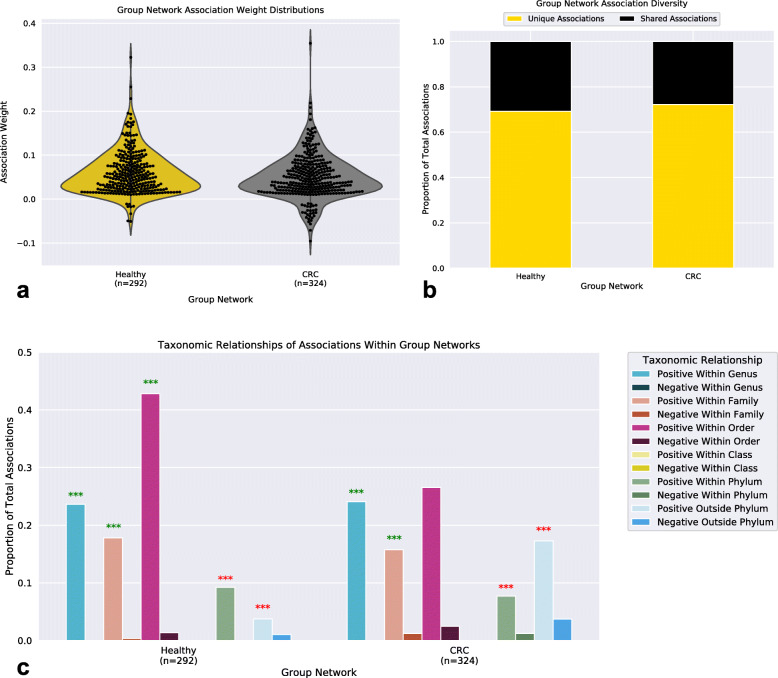
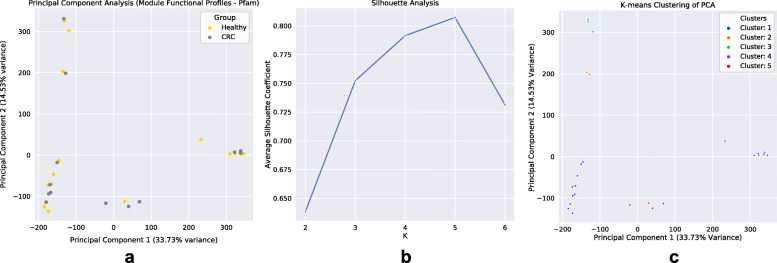
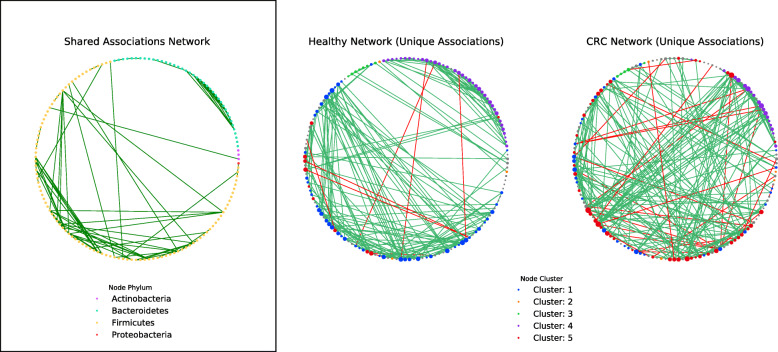
By applying a novel set of analyses on 252 previously published whole-genome shotgun sequenced fecal samples from healthy and late-stage colorectal cancer subjects, we identify taxonomic, functional, and structural changes within the cancer-associated human gut microbiome. Bacterial association networks constructed from these data exhibited widespread differences in the underlying bacterial community structure between healthy and colorectal cancer associated gut microbiomes. Within the cancer-associated ecosystem, bacterial species were found to form associations with other species that are taxonomically and functionally dissimilar to themselves, as well as form modules functionally geared towards potential changes in the tumor-associated ecosystem. Bacterial community profiling of these samples revealed a significant increase in species diversity within the cancer-associated gut microbiome, and an elevated relative abundance of species classified as originating from the oral microbiome including, but not limited to, Fusobacterium nucleatum, Peptostreptococcus stomatis, Gemella morbillorum, and Parvimonas micra. Differential abundance analyses of community functional capabilities revealed an elevation in functions linked to virulence factors and peptide degradation, and a reduction in functions involved in amino-acid biosynthesis within the colorectal cancer gut microbiome.
We utilize whole-genome shotgun sequenced fecal samples provided from a large cohort of late-stage colorectal cancer and healthy subjects to identify a number of potentially important taxonomic, functional, and structural alterations occurring within the colorectal cancer associated gut microbiome. Our analyses indicate that the cancer-associated ecosystem influences bacterial partner selection in the native microbiota, and we highlight specific oral bacteria and their associations as potentially relevant towards aiding tumor progression.
结直肠癌是全球癌症相关死亡的主要原因。人类肠道微生物组已成为研究理解结直肠癌的发生、发展和治疗的活跃领域。尽管多项研究发现结直肠癌患者肠道微生物组中特定细菌的携带存在显著改变,但没有一种细菌与所有病例都有明确关联。物种携带的改变是癌症形成的原因还是结果尚不清楚,但很明显的是,应该关注理解与癌症相关的肠道微生物组中细菌群落结构的变化。
通过对 252 个先前发表的来自健康和晚期结直肠癌患者的全基因组 shotgun 测序粪便样本应用一套新的分析方法,我们确定了与癌症相关的人类肠道微生物组中的分类学、功能和结构变化。从这些数据构建的细菌关联网络显示了健康和结直肠癌相关肠道微生物组之间基础细菌群落结构的广泛差异。在癌症相关的生态系统中,细菌物种与在分类学和功能上与自身不同的其他物种形成关联,并且形成了针对肿瘤相关生态系统中潜在变化的功能模块。对这些样本的细菌群落分析显示,癌症相关肠道微生物组中的物种多样性显著增加,并且来源于口腔微生物组的物种相对丰度升高,包括但不限于具核梭杆菌、口腔链球菌、牙龈卟啉单胞菌和微小消化链球菌。群落功能能力的差异丰度分析显示,与毒力因子和肽降解相关的功能升高,而与结直肠癌肠道微生物组中氨基酸生物合成相关的功能降低。
我们利用来自大量晚期结直肠癌和健康受试者的全基因组 shotgun 测序粪便样本,确定了在与结直肠癌相关的肠道微生物组中发生的一些潜在重要的分类学、功能和结构改变。我们的分析表明,癌症相关的生态系统影响了天然微生物群中细菌伙伴的选择,我们强调了特定的口腔细菌及其关联可能与辅助肿瘤进展有关。