Department of Computer Science and Information Engineering, National Chung Cheng University, Chiayi, Taiwan.
Department of Infectious Diseases, Taichung Veterans General Hospital, Taichung, Taiwan.
Genome Biol. 2021 Mar 31;22(1):95. doi: 10.1186/s13059-021-02282-6.
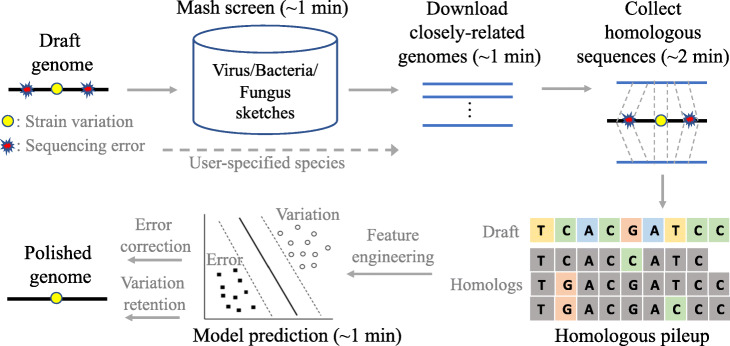
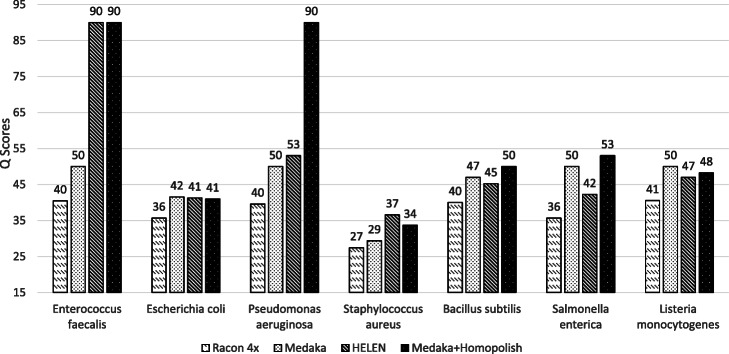
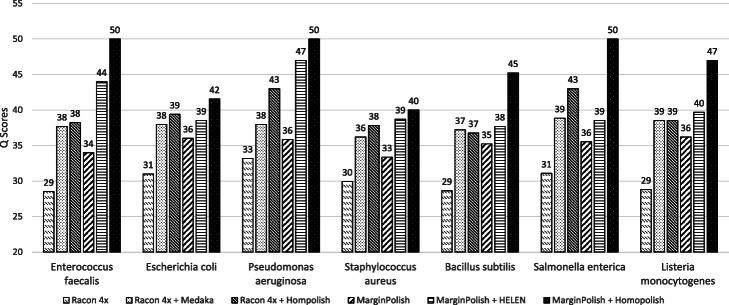
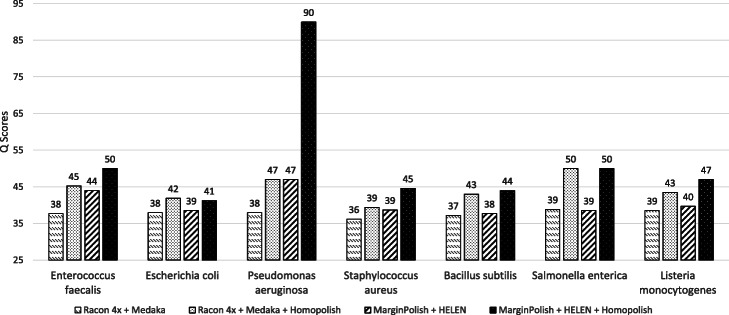
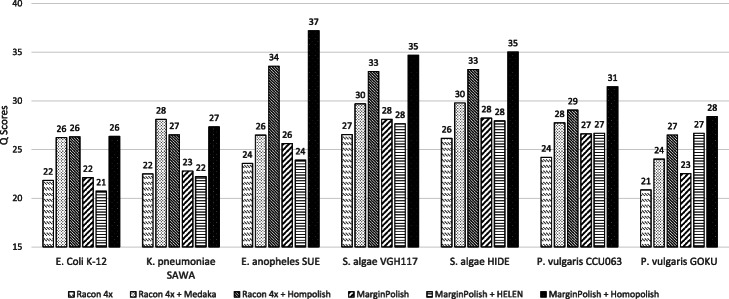
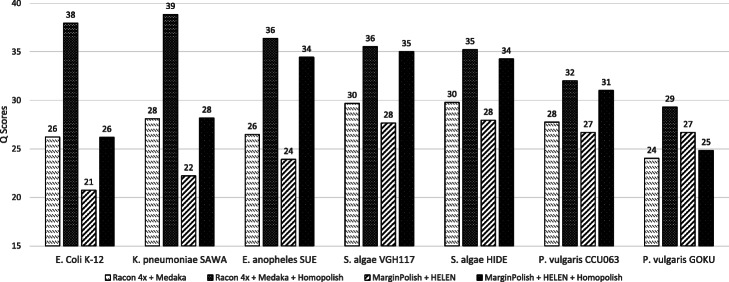
Nanopore sequencing has been widely used for the reconstruction of microbial genomes. Owing to higher error rates, errors on the genome are corrected via neural networks trained by Nanopore reads. However, the systematic errors usually remain uncorrected. This paper designs a model that is trained by homologous sequences for the correction of Nanopore systematic errors. The developed program, Homopolish, outperforms Medaka and HELEN in bacteria, viruses, fungi, and metagenomic datasets. When combined with Medaka/HELEN, the genome quality can exceed Q50 on R9.4 flow cells. We show that Nanopore-only sequencing can produce high-quality microbial genomes sufficient for downstream analysis.
纳米孔测序技术已广泛应用于微生物基因组的重建。由于错误率较高,基因组上的错误通过基于纳米孔读取数据训练的神经网络进行校正。然而,系统错误通常仍未得到校正。本文设计了一种利用同源序列进行纳米孔系统错误校正的模型。所开发的程序 Homopolish 在细菌、病毒、真菌和宏基因组数据集上优于 Medaka 和 HELEN。当与 Medaka/HELEN 结合使用时,基因组质量可以超过 R9.4 流动池上的 Q50。我们表明,仅使用纳米孔测序就可以生成足够用于下游分析的高质量微生物基因组。