Department of Chemistry and Biochemistry, University of California-Los Angeles, Los Angeles California 90095.
Departments of Molecular and Cell Biology and Plant and Microbial Biology, University of California-Berkeley, Berkeley, California 94720 and Environmental Genomics and Systems Biology, Lawrence Berkeley National Laboratory, Berkeley, CA 94720.
Plant Cell. 2021 May 31;33(4):1058-1082. doi: 10.1093/plcell/koab042.
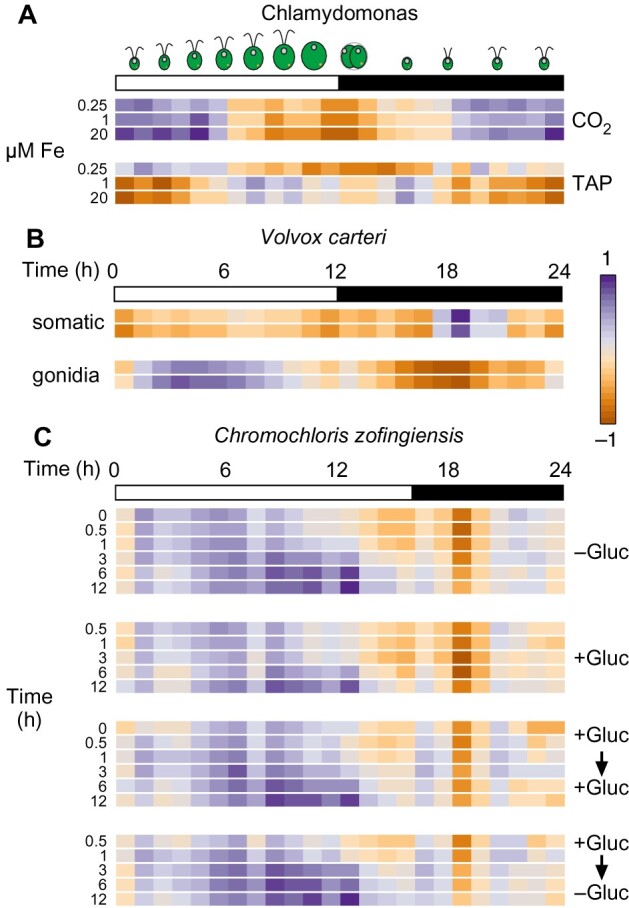
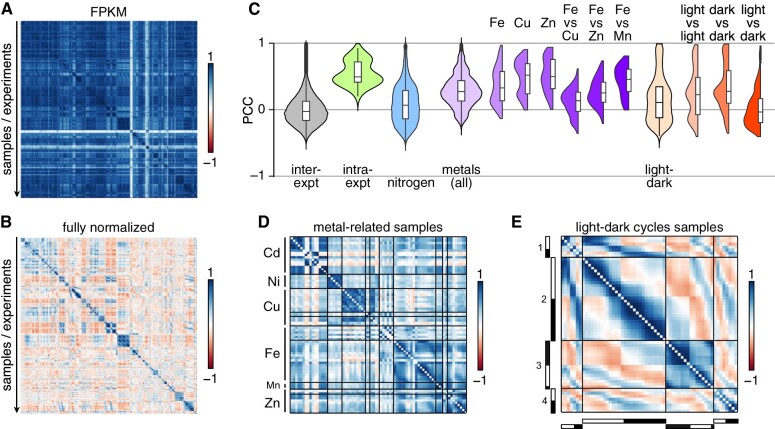
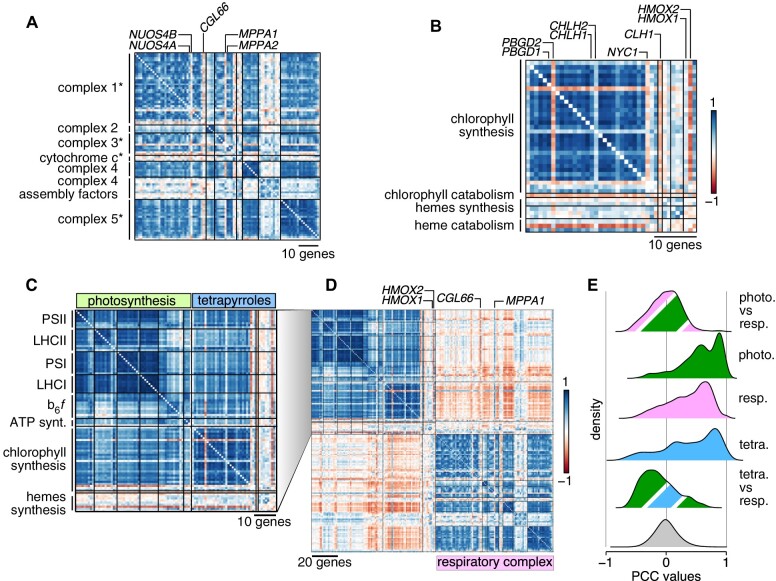
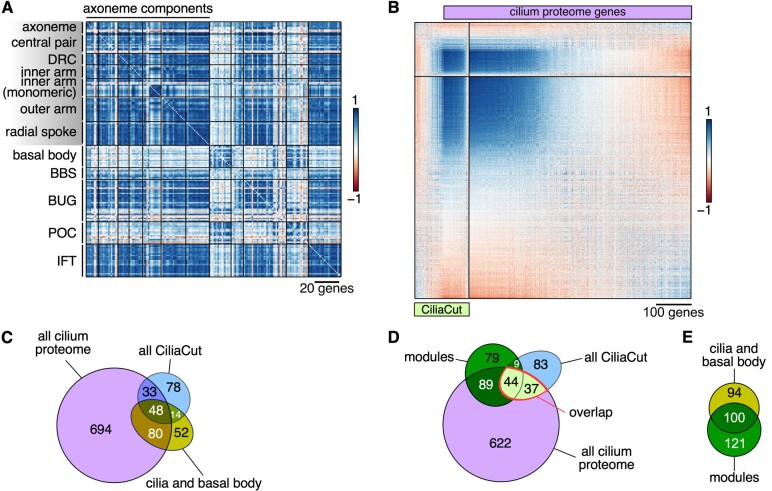
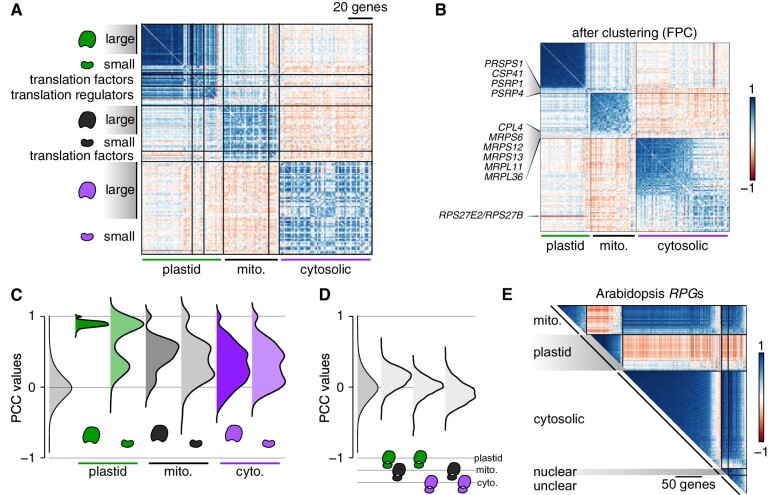
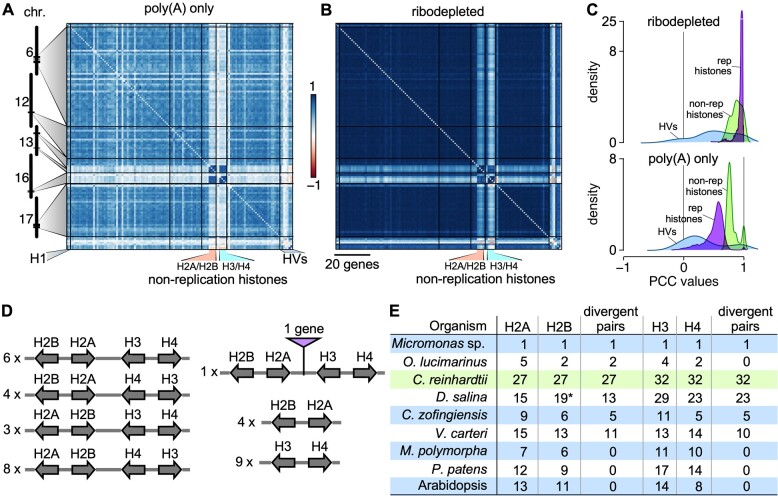
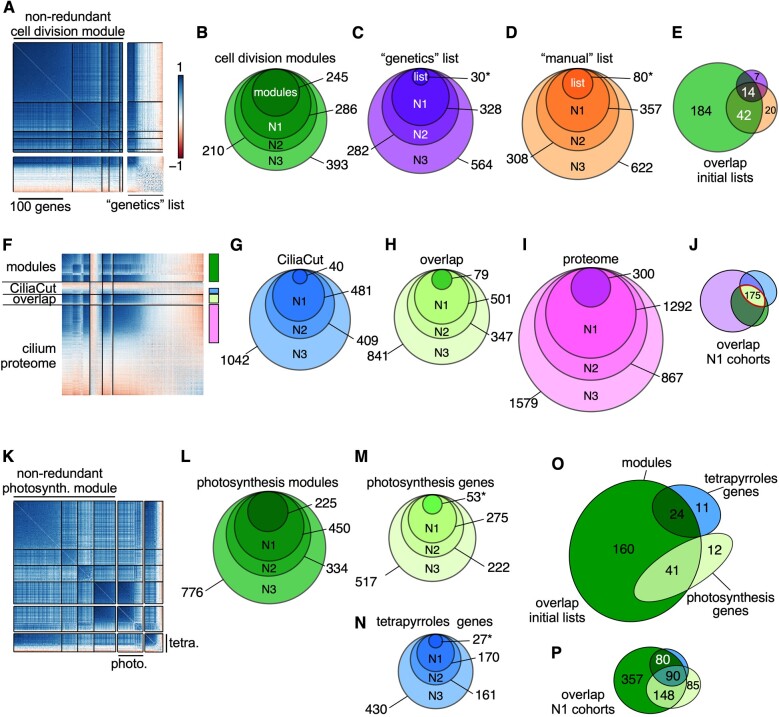
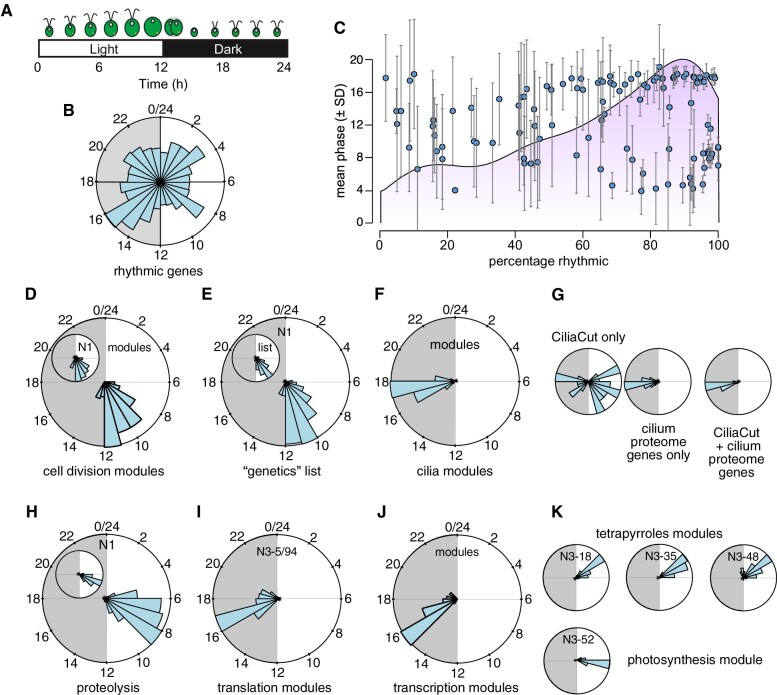
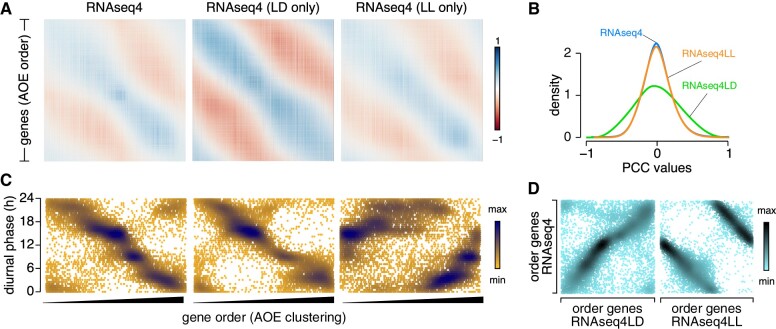
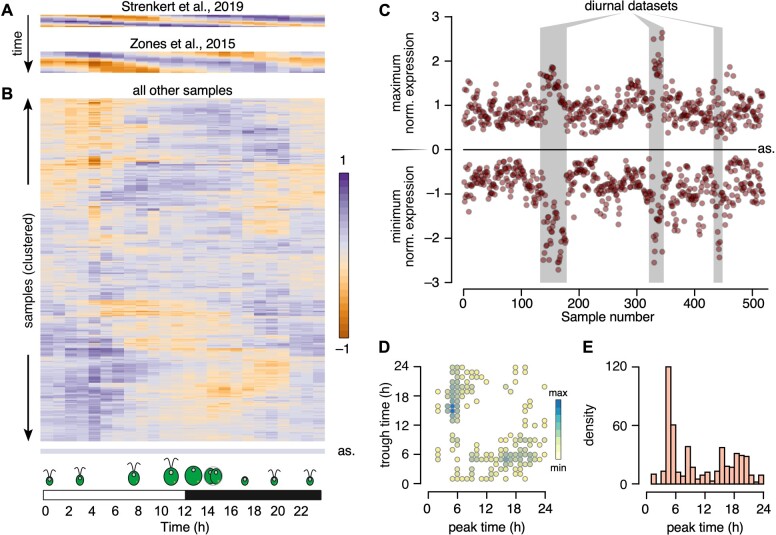
The unicellular green alga Chlamydomonas reinhardtii is a choice reference system for the study of photosynthesis and chloroplast metabolism, cilium assembly and function, lipid and starch metabolism, and metal homeostasis. Despite decades of research, the functions of thousands of genes remain largely unknown, and new approaches are needed to categorically assign genes to cellular pathways. Growing collections of transcriptome and proteome data now allow a systematic approach based on integrative co-expression analysis. We used a dataset comprising 518 deep transcriptome samples derived from 58 independent experiments to identify potential co-expression relationships between genes. We visualized co-expression potential with the R package corrplot, to easily assess co-expression and anti-correlation between genes. We extracted several hundred high-confidence genes at the intersection of multiple curated lists involved in cilia, cell division, and photosynthesis, illustrating the power of our method. Surprisingly, Chlamydomonas experiments retained a significant rhythmic component across the transcriptome, suggesting an underappreciated variable during sample collection, even in samples collected in constant light. Our results therefore document substantial residual synchronization in batch cultures, contrary to assumptions of asynchrony. We provide step-by-step protocols for the analysis of co-expression across transcriptome data sets from Chlamydomonas and other species to help foster gene function discovery.
单细胞绿藻莱茵衣藻是研究光合作用和叶绿体代谢、纤毛组装和功能、脂质和淀粉代谢以及金属动态平衡的首选参考系统。尽管经过几十年的研究,数千个基因的功能仍在很大程度上未知,需要新的方法来明确地将基因归类到细胞途径中。不断增加的转录组和蛋白质组数据集现在允许基于整合共表达分析的系统方法。我们使用了一个包含 518 个深度转录组样本的数据集,这些样本来自 58 个独立的实验,以确定基因之间潜在的共表达关系。我们使用 R 包 corrplot 可视化共表达潜力,以便轻松评估基因之间的共表达和反相关关系。我们从多个经过精心整理的与纤毛、细胞分裂和光合作用相关的列表的交集处提取了数百个高可信度基因,这说明了我们方法的强大功能。令人惊讶的是,即使在恒定光照下收集的样本中,莱茵衣藻实验在整个转录组中仍保留着显著的节律成分,这表明在样本收集过程中存在一个被低估的变量。因此,我们的结果记录了批量培养物中存在大量剩余同步,这与异步的假设相矛盾。我们提供了分析莱茵衣藻和其他物种转录组数据集中共表达的分步协议,以帮助促进基因功能发现。