Influenza Group, ICMR-National Institute of Virology, Pune, Maharashtra, India.
ICMR-National Institute of Virology, Pune, Maharashtra, India.
Indian J Med Res. 2021;153(1 & 2):166-174. doi: 10.4103/ijmr.IJMR_3418_20.
BACKGROUND & OBJECTIVES: Several phylogenetic classification systems have been devised to trace the viral lineages of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). However, inconsistency in the nomenclature limits uniformity in its epidemiological understanding. This study provides an integration of existing classifications and describes evolutionary trends of the SARS-CoV-2 strains circulating in India.
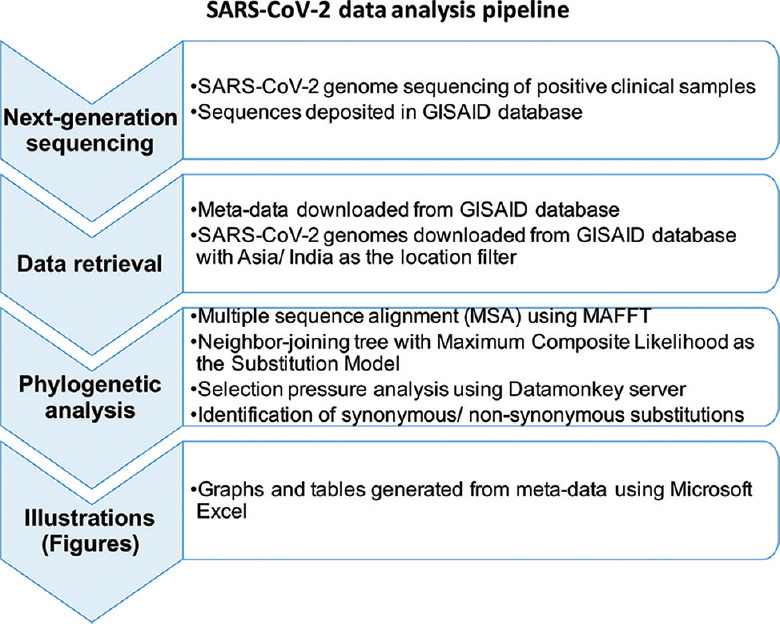
The whole genomes of 330 SARS-CoV-2 samples were sequenced using next-generation sequencing (NGS). Phylogenetic and sequence analysis of a total of 3014 Indian SARS-CoV-2 sequences from 20 different States/Union Territories (January to September 2020) from the Global Initiative on Sharing All Influenza Data (GISAID) database was performed to observe the clustering of Nextstrain and Phylogenetic Assignment of Named Global Outbreak LINeages (Pangolin) lineages with the GISAID clades. The identification of mutational sites under selection pressure was performed using Mixed Effects Model of Evolution and Single-Likelihood Ancestor Counting methods available in the Datamonkey server.
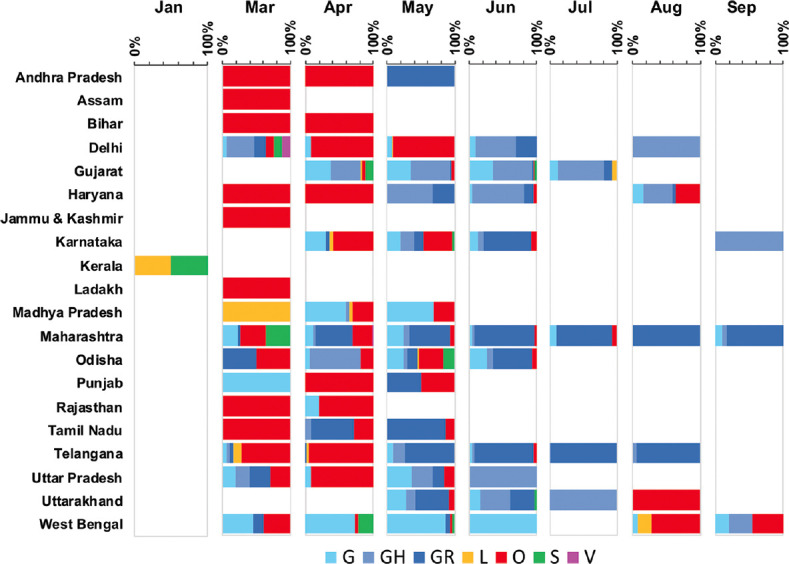
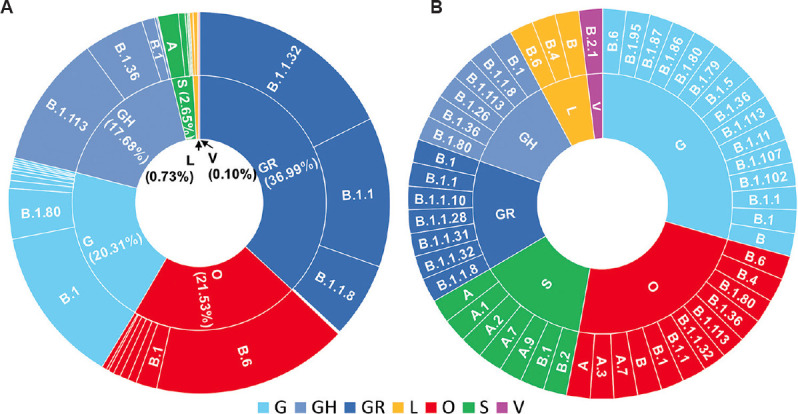
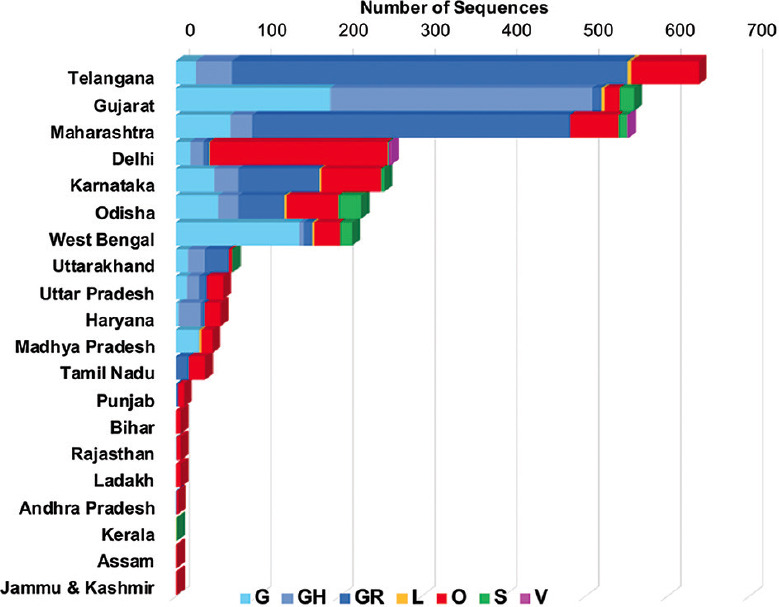
Temporal data of the Indian SARS-CoV-2 genomes revealed that except for Uttarakhand, West Bengal and Haryana that showed the circulation of GISAID clade O even after July 2020, the rest of the States showed a complete switch to GR/GH clades. Pangolin lineages B.1.1.8 and B.1.113 identified within GR and GH clades, respectively, were noted to be indigenous evolutions. Sites identified to be under positive selection pressure within these clades were found to occur majorly in the non-structural proteins coded by ORF1a and ORF1b.
INTERPRETATION & CONCLUSIONS: This study interpreted the geographical and temporal dominance of SARS-CoV-2 strains in India over a period of nine months based on the GISAID classification. An integration of the GISAID, Nextstrain and Pangolin classifications is also provided. The emergence of new lineages B.1.1.8 and B.1.113 was indicative of host-specific evolution of the SARS-CoV-2 strains in India. The hotspot mutations such as those driven by positive selection need to be further characterized.
已经设计了几种系统发育分类法来追踪严重急性呼吸系统综合征冠状病毒 2 (SARS-CoV-2) 的病毒谱系。然而,命名法的不一致限制了其流行病学理解的一致性。本研究整合了现有的分类,并描述了在印度流行的 SARS-CoV-2 株的进化趋势。
使用下一代测序 (NGS) 对 330 个 SARS-CoV-2 样本的全基因组进行测序。对来自全球流感共享倡议数据(GISAID)数据库的 20 个不同邦/联邦属地的 3014 个印度 SARS-CoV-2 序列(2020 年 1 月至 9 月)的总计 3014 个序列进行系统发育和序列分析,以观察 Nextstrain 和系统发育命名的全球暴发谱系(Phylogenetic Assignment of Named Global Outbreak LINeages, Pangolin)谱系与 GISAID 进化枝的聚类。使用 Datamonkey 服务器中提供的混合效应进化模型和单似然祖先计数方法,对选择压力下突变位点的鉴定进行了分析。
印度 SARS-CoV-2 基因组的时间数据表明,除了北阿坎德邦、西孟加拉邦和哈里亚纳邦在 2020 年 7 月后仍显示出 GISAID 进化枝 O 的流行外,其余邦都完全转向了 GR/GH 进化枝。在 GR 和 GH 进化枝中分别发现的 Pangolin 谱系 B.1.1.8 和 B.1.113 被认为是本土进化。在这些进化枝中鉴定为受到正选择压力的位点主要发生在 ORF1a 和 ORF1b 编码的非结构蛋白中。
本研究基于 GISAID 分类,解释了 SARS-CoV-2 株在印度九个月期间的地理和时间优势。还提供了 GISAID、Nextstrain 和 Pangolin 分类的整合。新谱系 B.1.1.8 和 B.1.113 的出现表明 SARS-CoV-2 株在印度的宿主特异性进化。需要进一步对热点突变(如受正选择驱动的突变)进行特征描述。